Oxígeno
Donald Canfield
Agradecimientos
Debo empezar por mostrar mi agradecimiento a todos mis buenos amigos y colegas que han trabajado duro de diversas formas para contribuir a esclarecer la dinámica del reciclado de oxígeno tanto en la Tierra moderna como en la antigua. Este libro es su historia tanto como la mía. El lector encontrará a la mayoría de ellos a medida que la narración se desarrolla, pero me gustaría destacar la inspiración que han supuesto Bob Berner, Tim Lenton, Rob Raiswell, John Hayes, Lee Kump, Penny Chisholm, Ed Delong, Nick Butterfield, Jorge Sarmiento, Osvaldo Ulloa, Bo Thamdrup, Bo Barker Jørgensen, Andrey Bekker, Bob Blankenship, Roger Buick, Fritz Widdel, Niels Peter Revsbech, Martin Brasier, Jake Waldbauer, Jochen Brochs, Birger Rasmussen, Bill Schopf, Paul Falkowski, Bill Martin, Dave Des Marais, John Waterbury, Sean Crowe, Simon Poulton, CarriAyne Jones, Jim Kasting, Minik Rosing, Christian Bjerrum, Tim Lyons, Ariel Anbar, Stefan Bengtson, Andy Knoll, Roger Summons, Dave Johnston, James Farquhar, Nick Lane, Jim Gehling, Guy Narbonne, Tais Dahl, Daniel Mills y Emma Hammarlund. También quiero reconocer el estímulo constante del grupo NordCEE, repartido entre la Universidad del Sur de Dinamarca, la Universidad de Copenhague y el Museo Sueco de Historia Natural. Muchos de los héroes de esta historia ya no se encuentran con nosotros, pero su inspiración continúa, y entre ellos están Dick Holland, Vladimir Vernadsky, Preston Cloud, Karl Turekian y Bob Garrels. Este libro ha progresado un poco a trompicones, pero estoy especialmente agradecido a la División de Ciencias Geológicas y Planetarias de Caltech, y sobre todo a mi anfitrión Woody Fischer, por conseguir una beca Moore para sostener dos meses de dicha productiva en compañía de mi familia y a salvo de las distracciones hogareñas. Durante el proceso de escritura, he recibido comentarios valiosos sobre capítulos concretos de Bob Blankenship, Minik Rosing, Bob Berner, Tais Dahl, Emma Hammarlund y Guy Narbonne. Estoy agradecido a Bill Martin y Lee Kump, que aportaron comentarios al texto completo, y especialmente a Raymond Cox, Tim Lyons y mi correctora Sheila Ann Dean, cuyos exhaustivos comentarios y correcciones han mejorado mucho el manuscrito. Gracias a mi editora, Alison Kalett, de Princeton University Press, tanto por su paciencia como por sus amplias críticas. Varias imágenes, o los datos para generarlas, fueron aportadas amablemente por Minik Rosing, Emma Hammarlund, James Farquhar, Matt Saltzan, Niels Peter Revsbech, Ken Williford, Martin van Kranendonk, Bruce Wilkenson, Bill Schopf, Tais Dahl, Eric Condliffe, Bo Thamdrup, Jakob Zopfi y Lawrence David. Por último, quiero reconocer el apoyo generoso de mis fuentes de financiación, entre ellas la Fundación Nacional de Investigación de Dinamarca (Danmarks Grundforskningsfond), el Consejo Europeo de Investigación (Oxygen Grant) y el Instituto Agouron.
Prefacio
Si es usted como yo, lo más probable es que no piense mucho en el aire que respira, a menos que huela mal por alguna razón. Sin embargo, nuestro aire es muy especial. Contiene un 21% de oxígeno, y nuestro mundo es el único, al menos que sepamos hasta ahora, con unos niveles tan elevados. Esto es bueno para nosotros, porque somos animales grandes y necesitamos mucho oxígeno para vivir. También lo precisan nuestros peludos amigos los perros y los gatos, así como vacas, pollos, ovejas, cerdos y otros animales en los que se basa gran parte de nuestra dieta. El oxígeno quema el combustible que calienta nuestras casas, y permite el brillo acogedor de una fogata en una noche fresca de otoño. Por expresarlo en una frase, el oxígeno es una marca distintiva de la Tierra; sus altos niveles en nuestra atmósfera definen el contorno de nuestra existencia y la naturaleza de la vida animal en la Tierra.
Dada la importancia del oxígeno en la Tierra, podemos considerar una serie de cuestiones. Por ejemplo, ¿de dónde viene todo ese oxígeno? ¿Por qué es tan alto su nivel? ¿Qué controla la concentración atmosférica de este importante gas? Después nos podemos preguntar si las concentraciones de oxígeno han sido siempre tan altas y, si no, cómo han cambiado en el tiempo, y por qué. Por último, y dada la importancia del oxígeno para la biosfera actual, ¿hay algún indicio de que la historia de los niveles del oxígeno pueda estar acoplada de alguna manera a la historia de la evolución biológica en la Tierra?
Este libro trata de la historia del oxígeno atmosférico en la Tierra, e intentaré responder esas preguntas en las siguientes páginas. Una de las conclusiones inevitables, que ofrezco aquí a modo de avance, es que el control del oxígeno es un fenómeno global, y que este gas persiste a alto nivel gracias a una fascinante interacción entre procesos biológicos y geológicos. La naturaleza de esa interacción ha cambiado con el tiempo, de lo que ha resultado una intensa historia de la evolución del oxígeno; esta historia, tal como la entendemos, se revelará en las páginas siguientes.
La trama versa también sobre la gente implicada en desentrañar la historia de la evolución del oxígeno. De hecho, comprender esa historia se ha vuelto una materia popular, y hay ahora muchos científicos implicados en su exploración. Muchos de estos investigadores son buenos amigos y colegas, y todos ellos han contribuido a una vida de trabajo maravillosa y fértil. También hay héroes en esta historia; pensadores visionarios que forjaron los caminos que otros siguieron, incluido yo mismo. Algunos de esos pensadores estuvieron décadas por delante de su tiempo.
El libro es también sobre cómo sabemos lo que sabemos. Presento las evidencias. Están basadas, sobre todo, en pistas dejadas en primitivas rocas sedimentarias. Parte de la evidencia es buena, y otra parte no lo es tanto, especialmente cuando miramos a rocas muy viejas en las que los estragos del tiempo se han cobrado su precio. La preservación del registro geológico, sin embargo, es parte de la historia, y debemos usar las evidencias que tenemos. Esto significa que a veces seremos incapaces de alcanzar conclusiones firmes. Las incertidumbres de este tipo son una parte esencial del proceso científico, y por tanto les dedico atención. Aun así, a menudo podemos mirar un problema con múltiples líneas de evidencia, y si aplicamos la navaja de Ockham
[1] podemos llegar normalmente a una hipótesis de trabajo razonable sobre el significado de los datos. También trato de subrayar los casos en que nuestras ideas han evolucionado a medida que los datos han mejorado, se han incrementado o se han comprendido mejor.
No todas las evidencias, sin embargo, vienen de la geología. El argumento tiene una fuerte componente biológica. En ocasiones tenemos que mirar a los organismos y ecosistemas modernos para ver cómo funcionan. Aportan pistas importantes para ayudarnos a entender cómo funcionaba el mundo antiguo, especialmente en detalles que el registro geológico no puede proveer fácilmente. También debemos considerar la evolución biológica. Por ejemplo, ¿cómo se originó la producción biológica de oxígeno? Esta es una historia fascinante.
A veces también necesitamos entender tópicos complejos, tales como el funcionamiento de la fotosíntesis, o cómo los isótopos se pueden utilizar para revelar la historia del oxígeno. Mi objetivo ha sido hacer estas discusiones accesibles a cualquier lector interesado en la ciencia, de modo que intento introducir los principios difíciles con el suficiente contexto para hacerlos ampliamente comprensibles. También uso notas al final del libro para explicar principios y procesos con el detalle que puede demandar un especialista u otro lector especialmente interesado en la materia. Mi esperanza, sin embargo, es que la historia no requiera realmente visitar las notas a menos que el lector quiera aprender más.
Por último, esta es una narración sobre el tiempo, sobre vastas extensiones de tiempo. El planeta Tierra tiene unos 4500 millones de años, que es más o menos un tercio de la edad del universo. Estudié química en la universidad, y mi experiencia con el tiempo, al menos científicamente, estaba limitada a las horas o días de una reacción química. Nuestras vidas enteras no son sino un parpadeo en comparación con la edad de la Tierra. En verdad, pensar sobre los inmensos senderos del tiempo geológico no me resultó nada fácil. La enormidad del tiempo geológico nos desafía a imaginar cómo los procesos lentos, como la evolución o la construcción de una montaña, pueden funcionar realmente. Ahora me siento más cómodo con el tiempo geológico, y con las escalas temporales de los procesos de la geología y la evolución, pero me compadezco con la dificultad de percibir procesos que se desarrollan en escalas de tiempo inmensamente mayores que la duración de la vida humana. En cualquier caso, la vastedad del tiempo geológico ya fue reconocida hace siglos, y hecha célebre en los comentarios finales de la obra clásica de 1788 Teoría de la Tierra, de James Hutton:
«El resultado, por tanto, de nuestra presente indagación es que no hallamos vestigio de un principio, ni expectativa de un final».
No mucho después de que se publicara el libro de Hutton, se hizo obvio que, en ciertos estratos de rocas, podían reconocerse distintas colecciones de fósiles. Esto resultaba de utilidad práctica para identificar estratos que pudieran tener interés económico, pero también resultó evidente que esas capas podían ser divididas, subdivididas y datadas unas respecto de otras. Un principio clave de la datación fue la simple deducción, hecha en el siglo XVII por el erudito danés Nicolaus Steno, de que un estrato de sedimento depositado sobre otro es más joven que el estrato de abajo. Esto se conoce como la ley de superposición.
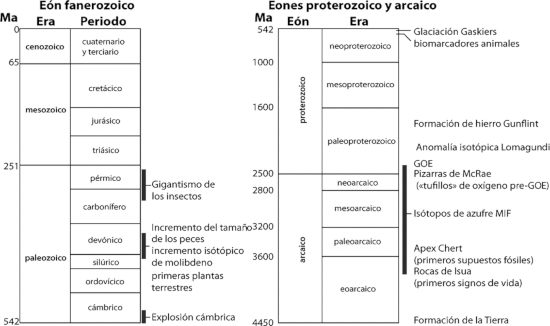
Escala del tiempo geológico con los principales acontecimientos que explica el texto. Escala de tiempo basada en Gradstein (2004).
Las divisiones principales se describían a menudo por la pérdida o aparición de grupos de fósiles distintivos, y correlacionando un afloramiento de terreno con otro; estas divisiones podían reconocerse de un lugar a otro, y finalmente alrededor de todo el mundo. Las divisiones recibieron nombres y, a medida que los métodos de datación por radioisótopos se fueron haciendo disponibles, las rocas se pudieron datar con precisión. Lo que resulta es la escala del tiempo geológico. Este es nuestro mapa de carreteras, nuestra vara de medir, y es tan central a la geología como la tabla periódica lo es a la química. Hay múltiples escalas en esas divisiones, que barren desde eones (de cientos de millones a miles de millones de años), eras (decenas a cientos de millones de años) y periodos (decenas de millones de años) hasta estadios (millones de años). Una versión abreviada de la escala del tiempo geológico se muestra en la tabla de la página anterior, que incluye algunos sucesos clave y lugares que se discuten en el texto.
Escribir este texto ha sido a la vez un gozo y una inmensa experiencia de aprendizaje. Ha sido muy divertido tratar de enfocar mi pensamiento en tópicos que han sido borrosos, y seguir el desarrollo histórico de las muchas ideas que se presentan aquí. El único aspecto negativo del proceso de escritura fue mi percepción súbita de que sólo podía discutir una fracción de la literatura pertinente sobre cualquier materia dada. Así pues me disculpo por anticipado ante mis colegas y amigos cuyo trabajo no he mencionado. No me ha pasado inadvertido. Pese a la necesidad de economizar en el texto, deseo que este libro represente una puesta al día de la materia, pero reconozco que es probable que dentro de 30 años se escriba otro libro bien distinto. Espero que disfruten.
DON CANFIELD (Odense, Dinamarca)
Capítulo 1
¿De qué trata el planeta Tierra
Voy sentado en el tren, como suelo hacer, viajando entre Odense y Copenhague. Acabamos de arrancar desde la parada de Ringsted. Miro por la ventana. La escena es típica del campo danés, mezcla de granjas y bosques. Hay vacas pastando perezosas en el prado, y más allá un granjero está cortando heno. Muy por encima, un halcón busca ratones en la hierba sin podar. Amo este paisaje. Me recuerda a los campos de Ohio donde crecí. No es espectacular, pero reconfortante y tranquilizador a su manera; un paisaje honrado poco propicio a la fanfarronada y el engaño. Entorno los ojos y el paisaje se funde en una masa verde, las vacas se vuelven fantasmas en la distancia. Los reabro y veo que pasamos junto a un área de bosque denso (o al menos lo que pasa por bosque en Dinamarca). Mi mente vaga y reflexiono sobre lo que veo. Dinamarca es un país pequeño y la tierra, incluido el bosque, está gestionada a fondo, de manera que la diversidad de la vida no es particularmente grande. Uno puede viajar a los bosques pluviales de Costa Rica o Brasil y sobrecogerse mucho más con los pájaros tropicales, las ranas, los insectos y la vegetación abundante. Aun así, incluso en Dinamarca, el paisaje es de un verde brillante y rebosante de vida. De hecho, la mires como la mires, la Tierra se caracteriza por la vida diversa y abundante. La cuestión que me preocupa ahora es: ¿por qué?
Se podría insinuar que toda la vida que vemos es simplemente la consecuencia de una larga historia de evolución biológica en la Tierra. En su maravilloso libro La vida en un planeta joven, mi colega y buen amigo Andy Knoll, de la Universidad de Harvard, documenta la cambiante cara de la vida durante los primeros 4000 millones de años de historia de la Tierra. Muestra cómo una variedad de innovaciones biológicas, como la invención de la fotosíntesis productora de oxígeno, por ejemplo, dieron forma fundamental a la historia de la vida. Después de que los organismos productores de oxígeno evolucionaran por vez primera, otros organismos que utilizan oxígeno les siguieron, y después prosperaron, se multiplicaron y evolucionaron hacia nuevas formas vivas también utilizadoras de oxígeno. Con el tiempo esto condujo a los animales, los organismos más complejos biológicamente de todos los que pueblan la Tierra. Sin oxígeno no habría animales. De manera que, claramente, las innovaciones durante la evolución biológica han dado forma, incluso definido, la biosfera. Pero ¿explica la evolución por sí sola la prodigalidad de la vida en nuestro planeta?
Para considerar esta cuestión, comparemos brevemente la Tierra con Marte. Los científicos todavía consideran posible que exista vida en Marte: al fin y al cabo, Marte tiene la misma edad que la Tierra y hay indicios de la presencia al menos ocasional de agua superficial y subterránea en el planeta. Mientras escribo esto, el rover Curiosity de la NASA está sondeando la superficie de Marte en busca de indicios de agua, y de pistas sobre cómo el agua interactúa con el entorno superficial del planeta. Como discutiremos con mayor detalle más abajo, y tal como sostiene la doctrina, donde hay agua puede haber vida. Y, sin embargo, si hay vida en Marte, no salta arriba y abajo como los Whos en Whoville gritando «¡estamos aquí, estamos aquí, estamos aquí!». En contraste, si unos exploradores intergalácticos sondearan la Tierra como nosotros hacemos ahora con Marte, resultaría imposible que la abundante vida de la Tierra les pasara inadvertida. La cuestión es, simplemente, ¿por qué hay tanta vida en la Tierra?
Para responder esto abandonaremos de momento las consideraciones evolutivas y empezaremos por una pregunta más fundamental: ¿cuáles son los ingredientes básicos que necesita la vida, o al menos la vida tal y como la conocemos? Mientras digiero mi almuerzo de sobras de lasaña, proclamo que la comida debe ser importante. Sin duda, pero no todos los organismos pueden comer lasaña, y me viene a la mente una clase entera de criaturas que no comen ningún tipo de materia orgánica en absoluto, sino que simplemente fabrican sus células a partir de sustancias inorgánicas. Es lo que hacen las plantas, que crecen a partir de dióxido de carbono y agua y usan la energía del Sol para combinar esos compuestos produciendo biomasa celular y oxígeno.
También lo hacen muchos otros tipos de organismos, y la mayoría de ellos no usan el Sol como fuente de energía. En vez de eso, consiguen su energía promoviendo la reacción entre sustancias inorgánicas en las llamadas reacciones de oxidación-reducción, donde los electrones son transferidos durante la reacción. Para investigar esta idea más a fondo, pensemos en la sal. Si pones sal en agua, se disuelve en una reacción que genera energía, pero los organismos no pueden crecer usando la energía de esa reacción; no se transfieren electrones; y los átomos de cloro y sodio tienen la misma carga en los cristales de sal que en la solución. Ahora pensemos en las vacas. Las vacas albergan enormes poblaciones de microbios en su sistema digestivo, y muchos de ellos generan metano. Muchos de estos microbios, los llamados metanógenos, crecen sin problemas combinando hidrógeno gaseoso y dióxido de carbono para formar gas metano. No se usa luz, pero los electrones son transferidos, y los metanógenos son felices, como seguramente lo son las vacas. Por lo tanto, una necesidad básica para la vida es la energía, que puede ser aportada por la luz o por una infinidad de reacciones de oxidación-reducción diferentes.
[2] Consideraremos estos asuntos con más detalle en el siguiente capítulo, pero de momento basta con subrayar que la energía es crucial para la vida.
La energía es indispensable, pero también necesitamos otras cosas. Las células están hechas de carbono, oxígeno, hidrógeno, nitrógeno, fósforo y azufre, como ingredientes principales, junto a un juego completo de metales minoritarios y otros elementos. Todos estos compuestos son cruciales para la construcción de componentes celulares básicos como la membrana celular, el material genético (ADN y ARN) y todas las proteínas y las demás moléculas que se usan para operar la maquinaria de la célula.
Otro ingrediente básico de la vida, al menos de la vida tal y como la conocemos, es un entorno acuoso estable. ¡La vida lo prefiere húmedo! Muchos organismos, desde luego, han evolucionado para vivir fuera de la esfera acuosa de nuestro planeta, pero aun así necesitan agua para vivir. Nosotros también, solo que la empaquetamos dentro de nuestro cuerpo. De manera que, ya estemos hablando de cactus del desierto, arañas, serpientes, árboles o de las bacterias más diminutas, todos necesitan agua. De hecho, esta es una de las razones, como ya mencionamos antes, de que la búsqueda de vida en nuestro sistema solar y más allá de él sea equivalente a la búsqueda de agua líquida. «Un momento», podría objetar el lector, «he oído que algunas bacterias y pequeñas algas viven en agua marina congelada e incluso en los glaciares en ciertos casos». Muy cierto, pero, si el organismo está vivo y creciendo,
[3] tiene acceso al agua líquida. En el caso del agua marina congelada, ello puede deberse a canales de salmuera formados excluyendo la sal del hielo en crecimiento; en los glaciares, la alta presión induce la fusión del hielo cerca del fondo, lo que aporta un entorno acuoso para los organismos. «Vale», podría añadir el lector, «pero he oído que la marca de temperatura para un organismo vivo ronda los 120 grados, muy por encima del punto de ebullición del agua en la superficie terrestre». Cierto de nuevo, pero esos organismos sólo se encuentran a altas presiones, como en las profundidades oceánicas donde el punto de ebullición del agua excede el límite superior de temperatura para la vida.
¿Qué es tan importante sobre el agua, en cualquier caso? Para empezar, el agua tiene propiedades especiales. Debido a su estructura física, una molécula de agua es bipolar, lo que significa que está ligeramente cargada, con una carga positiva en un lado y una carga negativa en el lado opuesto. Esta condición le permite disolver cualquiera de las llamadas sustancias químicas iónicas (también cargadas), muchas de las cuales constituyen los bloques de construcción de la vida. Estos incluyen nutrientes como el nitrato, el amonio y el fosfato, que dan lugar a componentes cruciales del ADN, el ARN y las membranas celulares, así como una plétora de otras sustancias que incluyen el sulfato y una variedad de metales minoritarios que contribuyen a construir la maquinaria bioquímica de la célula. El agua no sólo disuelve las sustancias, sino que estas sustancias también son transportadas por difusión y advección; y este movimiento proporciona un sistema por el que pueden ser distribuidas a las células. El agua también aporta el medio por el que los productos de desecho son exportados de la célula.
La naturaleza bipolar del agua también permite la formación de membranas celulares. Estas separan el ambiente externo del interior de la célula donde se gestiona el negocio de la célula. Las membranas celulares están hechas de moléculas especiales (fosfolípidos) en las que un extremo contiene grupos químicos amantes del agua (hidrofílicos) y el otro grupos químicos repelentes del agua (hidrofóbicos). Al formar una membrana, la parte amante del agua se coloca hacia fuera, mirando a la fase acuosa, mientras que la parte repelente del agua se coloca hacia dentro, cara a cara con otra fila de partes repelentes de agua cuyas partes afines al agua se disponen hacia afuera en la dirección opuesta. Esta bicapa de lípidos se une en círculo formando la membrana celular que separa el interior de la célula del medio externo. En conjunto, desde su capacidad para disolver y transportar los constituyentes químicos de la vida, hasta su capacidad de albergar estructuras de membrana, hacen del agua una sustancia química única.
O quizá estemos pensando con poca perspectiva, demasiado centrados en la Tierra. El agua es el fluido de la vida porque sus propiedades son perfectas para el tipo de vida que conocemos. Tal vez un tipo diferente de vida podría haber evolucionado en solventes diferentes con propiedades diferentes. Es difícil descartar esta posibilidad. A veces se mencionan potenciales solventes alternativos. Estos incluyen el amonio, el metano, el ácido sulfúrico o el fluoruro de hidrógeno (FH); a las temperaturas y presiones adecuadas, comparten algunas de las popiedades del agua, aunque no todas. Aparte de numerosos libros y películas de ciencia ficción, hay también una literatura científica muy activa sobre este tópico fascinante. Las discusiones sobre la vida en estas soluciones alternativas son, sin embargo, altamente especulativas, o incluso imaginativas, se podría decir. Por tanto, tomaré el camino fácil, y hasta donde podemos decir con certeza, el agua es el solvente único y perfecto para la vida.
En resumen, hemos subrayado tres ingredientes básicos para la vida. Son la energía, los componentes químicos que forman las células y el agua. Veremos a continuación que la disponibilidad de cada uno de ellos está vinculada por las propiedades especiales del planeta Tierra.
Empecemos por el agua. No es ningún secreto que la Tierra es un planeta acuoso. Desde las fascinantes imágenes de nuestro «planeta azul» tomadas por la NASA desde el espacio hasta la Rima del viejo marinero de Samuel Taylor Coleridge, todo nos recuerda la extensión sin límites de los océanos globales. No nos ocuparemos en detalle de por qué la Tierra tiene tanta agua —probablemente una combinación de la temprana pérdida de gases desde su interior y el suministro directo de los cometas—, sino más bien de por qué el agua que tenemos es, bueno, acuosa. La respuesta, por supuesto, es que la mayor parte del planeta tiene la temperatura adecuada, entre los puntos de ebullición y congelación del agua. Pero ¿por qué? En esto, al menos en parte, somos afortunados. Podemos pensar en ello de la siguiente forma. La Tierra se sitúa a cierta distancia del Sol dictada por su órbita. Y el Sol tiene cierto brillo dictado por su tamaño y composición química.
La cantidad de calor del Sol interceptada por la Tierra depende de una combinación de esos dos factores. Sin embargo, como todos los planetas de nuestro sistema solar se calientan por el mismo Sol, consideremos la distancia al Sol como la variable clave. Es fácil imaginar que si la Tierra estuviera más cerca del Sol recibiría más calor, y menos calor si estuviera más lejos. Resulta que la Tierra reside a una distancia del Sol tal que el calor es suficiente para permitir la persistencia del agua líquida. Cuando un planeta está más cerca del Sol, como Venus, la temperatura se vuelve demasiado alta, y el agua líquida hierve y se evapora a la atmósfera en el llamado «efecto invernadero desbocado» (
runaway greenhouse). Parte de esta agua puede incluso perderse por completo debido a procesos químicos en la estratosfera. Si el planeta está más lejos del Sol, como Marte, la superficie se vuelve demasiado fría y se congela. La zona situada a la distancia óptima del Sol (o de cualquier otra estrella, para el caso), donde puede persistir el agua líquida, se denomina «zona habitable», y a veces «zona Goldilocks».
[4]Pero la distancia al Sol es sólo parte de la historia. La Tierra tiene una atmósfera con gases de efecto invernadero que contribuyen a calentar la superficie. Sin ningún efecto invernadero, y con el albedo de la superficie tal y como es,
[5] la Tierra estaría congelada a unos 15 grados bajo cero. Por tanto, averiguar si un planeta está en la zona habitable es más enredado de lo arriba descrito. Ello requiere algunos cálculos bastante complejos sobre las transacciones de calor, que se empezaron a utilizar hace unas décadas; sin embargo, los modelos citados con más frecuencia fueron presentados en 1993 por Jim Kasting, de la Universidad Estatal de Pensilvania, junto con sus colaboradores Daniel Whitmire y Ray Raynolds. Jim ha sido un líder en aplicar su conocimiento detallado de la dinámica de la química atmosférica al entendimiento de la evolución tanto de la atmósfera terrestre como de la de otros planetas. Para abordar el asunto de la zona habitable Jim intentó, utilizando su modelo, mantener agua líquida en el planeta mediante cambios en los niveles de CO
2 (dióxido de carbono), ya que estos controlan el calentamiento por efecto invernadero. Es fácil imaginar que distintos niveles de CO
2 atmosférico serían necesarios para mantener la zona habitable en respuesta a diferencias de la luminosidad solar, que es básicamente la intensidad de la estrella, o del Sol en nuestro caso.
Con el modelo de Jim, el alcance exterior de la zona habitable se encuentra cuando las concentraciones de CO
2 atmosférico se vuelven tan altas que se forman nubes de CO
2. Estas nubes impiden que la radiación solar alcance la superficie del planeta, y por tanto incrementan el albedo planetario. El resultado final es un planeta congelado. Había otras consideraciones en el modelo de Jim que no trataré aquí, pero en definitiva Jim y sus colegas concluyeron que Marte queda probablemente justo fuera de la zona habitable. Por su parte, Venus queda también fuera de la zona habitable. En este caso, la luminosidad solar es simplemente demasiado alta. Incluso con unos niveles minúsculos de CO
2 atmosférico que aportan un mínimo calentamiento por efecto invernadero, la superficie del planeta se hace tan caliente que el agua se evapora a la atmósfera. Esta situación genera un efecto invernadero desbocado y unas temperaturas superficiales muy altas, ya que el agua es también un buen gas de efecto invernadero (¡y el más importante en la Tierra actual!).
[6] Según algunos cálculos de Jim, la frontera interior de la zona habitable puede situarse tan cerca como al 95% de la distancia del Sol a la Tierra. Esto es más o menos 7,2 millones de kilómetros más cerca del Sol de lo que estamos. Los resultados de los cálculos de Jim se presentan en la figura 1.1, y se tomen como se tomen significan que somos afortunados; la Tierra se sitúa ceñidamente en la zona habitable del Sol.
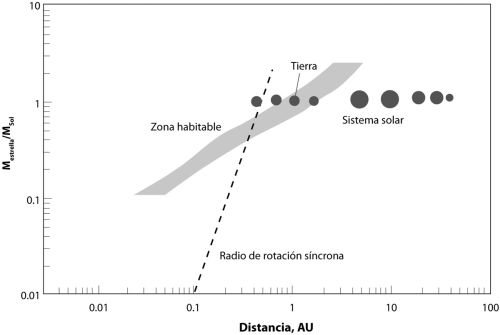
FIGURA 1.1 Zona de habitabilidad, según determinaron Jim Kasting y sus colegas. Se muestra la posición de los ocho planetas (más Plutón) de nuestro sistema solar. Una UA (unidad astronómica) es la distancia de la Tierra al Sol. El eje vertical muestra la proporción entre la masa de una estrella y la masa del Sol. A distancias de una estrella menores del radio de rotación síncrona, los planetas resultan bloqueados en unas rotaciones alrededor de su eje que multiplican exactamente (por números enteros pequeños como 1, 2, 3, o por sus fracciones como 3/2) el tiempo de rotación alrededor de la estrella (Mercurio rota 3 veces sobre su eje por cada 2 órbitas alrededor del Sol). En algunos casos, un planeta puede rotar una sola vez por cada periodo orbital, con la misma cara del planeta siempre mirando a la estrella. Los planetas que ocupan la zona habitable de una estrella pequeña están dentro del radio de rotación síncrona.
Si esto es cierto, ¿por qué seguimos contemplando la posibilidad de vida en Marte? En concordancia con los argumentos de Jim sobre la zona habitable, no hay evidencia de agua superficial permanente en Marte, al menos por ahora. Pero durante décadas de exploración por satélite y en superficie, incluidos los recientes y muy exitosos rovers Spirit y Opportunity de la Misión de Exploración por Rover (MER en sus siglas inglesas) y el sistema de imagen térmica de alta resolución THEMIS a bordo de la nave Mars Odyssey, el agua ha fluido y todavía fluye ocasionalmente en Marte. Las evidencias incluyen toda clase de canales, acequias, charcos y rocas sedimentarias cuya formación se puede explicar por la acción del agua. De hecho, el rover Curiosity ha aterrizado recientemente en la superficie de Marte y, mientras escribo esto, está explorando los alrededores de su lugar de aterrizaje, ¡que parece ser un antiguo lecho fluvial! Todo ello sumado a observaciones espectroscópicas de agua justo en y debajo de la superficie del suelo. Así, Marte demuestra que el agua líquida puede encontrarse, al menos ocasionalmente, un poco por fuera de la zona habitable. En contraste con la Tierra, sin embargo, cualquier vida que pueda haber en Marte, si es que existe, no es evidente y estaría aparentemente restringida en su ocurrencia y abundancia. Por tanto, Marte no sostiene ni puede sostener la magnitud de vida que encontramos en nuestro planeta.
Enterrada en la discusión sobre los cálculos de la zona habitable de Jim Kasting se halla la idea de que, a largas escalas de tiempo, la Tierra realmente regula su propia temperatura. Esta idea fue avanzada por primera vez por el cosmólogo Carl Sagan. Sagan hizo una gran contribución a nuestra comprensión de la composición de las atmósferas planetarias, y ayudó a enmarcar la discusión sobre la búsqueda de vida en el universo. Fue una enorme inspiración para las personas interesadas en la ciencia mediante su programa
Cosmos de la PBS (Public Broadcasting System), emitido originalmente en 1980. De mayor importancia aquí, sin embargo, él y su colega George Mullen se preguntaron por qué la Tierra no se congeló al principio de su historia, cuando el Sol era mucho menos luminoso que hoy.
[7] Las evidencias geológicas apuntan a la presencia más o menos continua de agua líquida desde hace tanto como 4200 millones de años. Sin embargo, con la abundancia actual de gases de efecto invernadero en la atmósfera terrestre, el planeta se debería haber congelado bajo la escasa luminosidad del joven Sol. Esto se conoce como «la paradoja del joven Sol débil». Sagan y Mullen arguyeron que esa paradoja podría resolverse con una alta concentración de gases de efecto invernadero como amonio y metano; estos gases son inestables en nuestra atmósfera oxigenada actual, pero podrían haber estado presentes en la atmósfera pobre en oxígeno de la Tierra temprana. Poco después se puntualizó, no obstante, que el amonio habría sido inestable fotoquímicamente, incluso en una atmósfera libre de oxígeno. Esto generó un grave problema para el modelo. Sin embargo, en un verdadero salto cuántico intelectual, Jim Walker, Paul Hays y Jim Kasting percibieron que el CO
2 podría muy bien haber sido el gas de efecto invernadero que evitara la congelación de aquella joven Tierra. Muy bien, CO
2 entonces. Pero esta propuesta va mucho más allá, porque Walker, Hays y Kasting también demostraron un mecanismo que regula realmente la temperatura superficial.
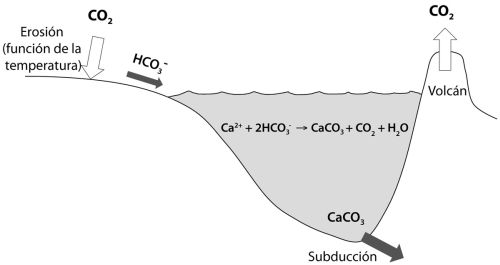
FIGURA 1.2 El ciclo del carbono actuando como regulador de la temperatura de la superficie de la Tierra. Redibujado a partir de Karting (2010).
La lógica es la siguiente. El dióxido de carbono es introducido constantemente desde el interior de la Tierra hacia la atmósfera. Ese CO
2 viene de los volcanes y de las fumarolas hidrotermales del fondo del océano. Sin embargo, si miramos con atención, vemos que esas fuentes de CO
2, o al menos la mayoría de ellas, se originan como resultado de la continua agitación de la Tierra en un proceso conocido como «tectónica de placas». En la práctica, la pérdida de calor desde el interior de la Tierra (estimada en unos 5000 grados en la zona media) causa que el manto (la capa situada inmediatamente debajo de la corteza terrestre) se mueva y se mezcle en el proceso denominado convección. Esta convección crea regiones de efusión volcánica, sobre todo en los océanos, que dividen la corteza terrestre en una serie de placas móviles que cabalgan sobre el manto subyacente. A medida que este proceso forma nuevo suelo oceánico, el suelo viejo va inyectándose de vuelta al interior del manto en el proceso denominado «subducción» (véase la figura 1.2). Este es un proceso violento que genera la mayoría de los grandes terremotos, y es el principal constructor de las cadenas montañosas. De modo que el CO
2 es liberado a la atmósfera, pero no se acumula allí para siempre. De hecho, es retirado activamente por un proceso conocido como «erosión química», donde el CO
2 reacciona con las rocas de la superficie terrestre.
[8] Un aspecto particularmente interesante de esta erosión es su sensibilidad a la temperatura: se acelera a temperaturas altas.
Con esto en mente, podemos empezar a imaginar cómo funciona la regulación de la temperatura a escala planetaria. Si la temperatura de la atmósfera se hace demasiado alta por alguna razón, la velocidad de la erosión se incrementará, y el CO2 será retirado más activamente de la atmósfera. La remoción incrementada de CO2 causará a su vez una caída de la concentración de CO2 en la atmósfera, reducirá el calentamiento por efecto invernadero y, como resultado, la temperatura bajará. Por lo tanto, se alcanza un punto de equilibrio entre la concentración de CO2, la temperatura y la tasa de remoción del CO2 por erosión. Supongamos que por alguna razón la Tierra se congela por completo. Esto puede haber ocurrido unas pocas veces a lo largo de la historia de la Tierra. Si ocurre, no tenemos que preocuparnos, al menos si consideramos grandes escalas de tiempo geológico. Los procesos tectónicos aseguran que el CO2 será añadido continuamente a la atmósfera. Sin agua líquida, no habrá retirada de CO2 por erosión, de modo que la concentración de CO2 seguirá aumentando hasta que las temperaturas alcancen el punto en que el hielo se funde, y la erosión comenzará de nuevo.
Durante la erosión, el CO2 se convierte en un ión soluble llamado bicarbonato (HCO3—), que precipita en forma de minerales como la calcita y la dolomita (como conchas de moluscos y arrecifes de coral, por ejemplo) en los océanos. Estos minerales se descomponen para dar de nuevo CO2 durante los procesos de subducción, lo que completa el ciclo. Resumiendo, por tanto, a través del ciclo de las rocas, la Tierra tiene un mecanismo activo de control de la temperatura, operado por los movimientos del manto y los procesos asociados de la tectónica de placas. Por tanto, la tectónica de placas es también crucial para permitir a la Tierra disfrutar de un suministro continuo de agua líquida a lo largo de la mayor parte de su larga historia.
Esta es una historia preciosa, pero ¿es cierta? Yo creo que debe serlo, al menos a grandes rasgos. Algunas evidencias geológicas, sin embargo, apuntan a concentraciones de CO
2 en la Tierra primitiva demasiado bajas como para calentar una Tierra iluminada por aquel Sol poco poderoso.
[9] De nuevo Jim Kasting ha entrado en la discusión remontándose a Sagan y Mullen, al sugerir que el metano puede haber sido un importante gas de efecto invernadero en la historia temprana de la Tierra. Esto ayudaría a explicar las bajas concentraciones de CO
2.
[10] Puede ser cierto, pero el ciclo del metano por sí mismo no conduce a un control de temperatura robusto de forma tan obvia como el CO
2. Muy recientemente, Minik Rosing y sus colegas (encontraremos a Minik de nuevo en el capítulo 7) han argumentado que tal vez hayamos estado enfocando el problema de manera incorrecta. Sugieren, de hecho, que tal vez el albedo de la Tierra joven era mucho más bajo que hoy,
[11] de modo que quizá no fuera necesario tanto gas de efecto invernadero para calentar el planeta. A Jim Kasting no le vuelve loco esta idea, pero unas concentraciones más bajas de CO
2 atmosférico pueden satisfacer tanto las evidencias geológicas sobre los niveles antiguos de CO
2 y producir el suficiente efecto invernadero para calentar el planeta en presencia de un débil Sol joven. Por lo tanto, el mecanismo de control por CO
2 tal y como fue descrito originalmente por Walker y Kasting puede todavía funcionar para regular la temperatura de la Tierra a lo largo del tiempo, incluso si los niveles primitivos de CO
2 fueron menores de lo que pensábamos.
Ahora volvamos a nuestra cuestión original. Una cosa es tener agua, y otra muy distinta sustentar una biosfera abundante. Como se mencionó al principio de este capítulo, la vida está por casi todas partes en la superficie de la Tierra. Pero ¿cómo la sustenta nuestro planeta? Intentemos hacer algunos cálculos. La vida fotosintética en la Tierra, funcionando a las presentes tasas de fotosíntesis, consumirían todo el CO
2 de la atmósfera en nueve años.
[12] Asimismo, la vida fotosintética de los océanos consumiría todo el fósforo disponible, un nutriente clave para las plantas acuáticas y las algas, en sólo 86 años.
[13] Siendo así, ¿cómo podemos sustentar tanta vida durante largas escalas de tiempo? Parte de la respuesta es que la mayor parte del CO
2 y de los nutrientes vinculados a las plantas y algas son liberados de vuelta a la atmósfera a medida que estos organismos mueren y son consumidos y descompuestos por toda clase de criaturas desde pandas gigantes hasta bacterias. Bien, pero aun así hay parte del material de las plantas y del fósforo que no se devuelve al entorno, sino que se entierra en sedimentos y acaba formando parte de las rocas. Si rehacemos nuestros cálculos para tomar en cuenta estas tasas de pérdida, encontramos que el CO
2 sería consumido en 13 000 años,
[14] y el fósforo en 29 000 años. Estas son todavía unas escalas de tiempo comparadas con los miles de millones de años que la vida ha perdurado en el planeta y los cientos de millones de años en que las plantas y los animales han poblado tierra firme. ¿Cómo explicamos esto?
La respuesta es realmente muy simple. Recurrimos a los mismos procesos tectónicos que usamos para explicar el papel del CO2 cuando resolvimos la paradoja del débil Sol joven. Afortunadamente, cuando los materiales son secuestrados en sedimentos marinos en la Tierra, no se quedan atrapados allí permanentemente. Los movimientos tectónicos del planeta así lo aseguran. Mediante los procesos de subducción, levantamiento de montañas y cambio del nivel del mar (el nivel del mar es influido tanto por la tectónica como por el clima), la mayoría de esos materiales acaban exponiéndose de nuevo al entorno erosivo. Durante la erosión, la materia orgánica vuelve a convertirse en CO2, el fósforo se libera de nuevo como solución, y una plétora de otros ingredientes para la vida vuelve a estar disponible para sustentar el crecimiento de los organismos. La clave aquí es que la magnitud de vida que disfrutamos en la Tierra es posible por el reciclaje activo de los constituyentes de la vida por procesos tectónicos. Esto fue reconocido por primera vez hace más de 200 años por James Hutton, a quien ya encontramos en el prefacio. Escribió en su tratado Teoría de la Tierra (1788):
El fin de la naturaleza al situar un fuego interno o poder de calor, y una fuerza de expansión irresistible, en el cuerpo de esta Tierra, es consolidar el sedimento recolectado en el fondo del mar, y formar de allí una masa de tierra permanente por encima del nivel del océano para el mantenimiento de plantas y animales.
Por último, ¿qué hay de la energía? Diré mucho más sobre la energía en el siguiente capítulo, sobre todo acerca de los tipos de energía necesarios para la vida, en muchos de los cuales no pensamos normalmente. En la Tierra moderna, sin embargo, la mayoría (probablemente más del 99%) de la energía de la biosfera viene en último término del Sol, alimentando la fotosíntesis de las plantas, las algas y los microbios (conocidos como cianobacterias; veremos mucho más sobre ellos en capítulos posteriores) que producen materia orgánica y oxígeno. Estos productos de la fotosíntesis son recombinados biológicamente en las grandes cadenas alimenticias de la Tierra. Por ejemplo, los copépodos del océano comen algas, los peces pequeños se comen a los copépodos, los peces grandes se comen a los pequeños, y peces aún más grandes se comen a aquellos. Estos peces mueren y se descomponen por la acción de una variedad de bacterias, que a su vez son consumidas por otros organismos. La cadena sigue y sigue, pero es alimentada, en último término, por la materia orgánica y el oxígeno producidos por la fotosíntesis. Como se describió más arriba, sin embargo, los organismos que producen el oxígeno y alimentan la biosfera obtienen sus bloques de construcción de material reciclado mediante la tectónica de placas. Por tanto, mientras que el Sol aporta la energía, la tasa a la que la tectónica recicla los componentes biológicos básicos establece el tempo.
Visto todo, debemos admitir que la Tierra es un lugar maravilloso para la vida. Se sitúa cómodamente dentro de la zona habitable del Sol. Además, su activa tectónica controla la temperatura del entorno de superficie, al aportar un suministro continuo de agua líquida, y también recicla los componentes básicos requeridos para estimular una vida abundante. Como veremos en el siguiente capítulo, la misma tectónica puede también haber generado unas condiciones óptimas para la primera biosfera.
Capítulo 2
La vida antes del oxígeno
Fue sin duda el viaje de mi vida. «¿Le produce claustrofobia?», preguntó el hombre. «No, en absoluto», mentí.
[15] «Bien», replicó él, «y haga lo que haga, no toque la manivela roja; sólo se usa en caso de emergencia». Con unas pocas instrucciones más, cerraron la escotilla y nos despegamos de la grúa. Nos dejaron meneándonos libremente sobre las olas, y esperé anticipando nuestro descenso.
Estaba sentado en el Alvin, el primer sumergible norteamericano de alta profundidad. Conmigo estaba mi buen amigo y colega Bo Barker Jorgensen, ahora en la Universidad de Aarhus en Dinamarca, y nuestro piloto Jim. Alvin fue utilizado por primera vez en 1964, y ha sido el principal vehículo para los descubrimientos en las profundidades del mar durante décadas desde entonces. Una entregada tripulación de apoyo mantiene a Alvin en perfectas condiciones y, según me dijeron, no es probable que quede una sola pieza del vehículo original. Pese a ello, una vez dentro de Alvin (al menos en 1999), uno no puede dejar de recordar la edad dorada de la exploración espacial, con interruptores de palanca y luces indicadoras incandescentes protegidas por esferitas de cristal. Te sientes como si fueras sentado en el módulo lunar, con una tecnología robusta que funciona. Y Alvin no sólo carece de sofisticación, sino también de amenidades. Los tres ocupantes están apretujados en una esfera de titanio de dos metros de diámetro, con los científicos sentados en extremos opuestos en unas alfombrillas de espuma. Hay que estirar el cuello como un pollo para mirar por los dos pequeños visores, que apuntan hacia abajo, o alternativamente uno puede fijar los ojos en los monitores de vídeo situados arriba. El calor corporal de los ocupantes calienta el batiscafo. Hay un tanque de oxígeno para suministrar este gas precioso, y un cartucho para retirar el CO2 que se acumula en el aire. No pregunten por el baño.
Estábamos flotando unos 1500 metros por encima del suelo oceánico de la cuenca de Guaymas en el golfo de California. Atravesando el golfo de California hay una zona de expansión conocida como la Elevación del Pacífico Este, que separa la placa norteamericana, al este, de la placa del Pacífico, al oeste. La divergencia de estas placas empuja lentamente la Baja Península apartándola del México continental. Este centro de expansión es algo inusual porque incluye una cubierta de sedimento espeso de cerca de un kilómetro depositada a lo largo de millones de años a partir de partículas suministradas por el antaño caudaloso río Colorado.
[16] El agua marina circula a través de rocas calientes del centro de propagación, formando fluidos hidrotermales que difunden hacia arriba a través del sedimento y emergen al suelo marino, precipitando grandes cantidades de yeso (CaSO
4×2H
2O) y suministrando abundante sulfuro al entorno local.
Empezamos nuestro descenso por la columna de agua. Mi nariz está pegada al puerto de observación mientras pasamos lentamente a través de la zona superior iluminada del océano.
[17] La luz desaparece en la negrura, y veo el destello luminescente ocasional de una criatura marina sin identificar. Pronunciamos pocas palabras, pero ninguna es necesaria mientras nos hundimos a través de la oscuridad con las notas de Brahms sonando en el reproductor de cintas suministrado por nuestro piloto. Después de una hora o así, las luces exteriores de
Alvin se encienden, y pestañeo al ver el más fantasmal de los paisajes. Grandes montículos de gusanos tubulares
Riftia se elevan de las sombras,
[18] meciéndose suavemente en colinas de yeso en expansión. Estos elegantes animales marinos, tan bellos en vida, no tienen boca ni ano, y viven a base de cultivar bacterias oxidantes de sulfuro en su intestino.
Riftia ha desarrollado un elaborado mecanismo para transportar oxígeno y sulfuro a las bacterias, que sobreviven combinando esas sustancias. Si miramos con atención, lo que parece ser nieve cayendo lentamente en la corteza de yeso es realmente una población de bacterias de vida libre oxidantes de sulfuro del género
Beggiatoa (lámina 1). Se extienden hasta el límite de las luces del
Alvin. También se ven muchos otros animales que, de un modo u otro, viven de la abundancia de vida microbiana sustentada por el sulfuro que emana de las soluciones hidrotermales. Como un recordatorio de esto, vemos alrededor de nosotros la efervescencia de las aguas hidrotermales calientes y ricas en sulfuro filtrándose desde la corteza que se acumula. El sulfuro alimenta a las bacterias que alimentan a los animales.
La vida abundante que encontramos en la cuenca de Guaymas y muchos otros sistemas hidrotermales de las profundidades oceánicas alrededor del mundo es alimentada por el sulfuro de las fumarolas, pero también requiere oxígeno de manera crucial. Estas bacterias oxidantes de sulfuro viven combinando el sulfuro con el oxígeno. Si quitáramos el oxígeno, ¿con qué nos quedaríamos? Los animales desaparecerían, las bacterias oxidantes de sulfuro no podrían sobrevivir, y casi todos los signos de vida que dominan nuestra visión desde las ventanillas del Alvin estarían ausentes. ¿Y qué ocurriría a escala planetaria? En el capítulo anterior vimos que, en la Tierra actual, más del 99% de la energía que alimenta a la biosfera viene del Sol canalizada a través de los fotosintetizadores productores de oxígeno. Si quitáramos los productores de oxígeno y toda la comida que generan, las grandes cadenas alimentarias de la Tierra se colapsarían dejando, bueno, ¿qué? Esta cuestión se hace pertinente cuando buscamos comprender la naturaleza de la vida en la Tierra antigua, antes de que evolucionara la producción de oxígeno.
Para responder esto, volvamos a la cuenca de Guaymas (o a casi cualquier otro centro de expansión con emisiones hidrotermales). Si quitáramos el oxígeno, la vida resultaría gravemente disminuida, pero aún quedaría alguna que podríamos encontrar. Pensemos en lo que suministran los fluidos hidrotermales. Como dije más arriba, en las profundidades de los océanos libres de oxígeno, el sulfuro sería de poca utilidad para la vida, pero los fluidos hidrotermales también contienen muchos otros compuestos, algunos de los cuales son muy interesantes para los organismos. Empecemos por el gas hidrógeno y el CO
2, que pueden alcanzar unas concentraciones bastante altas en los fluidos de las fumarolas hidrotermales. ¿Recuerdan las vacas del último capítulo? El mismo tipo de metanógenos (autótrofos)
[19] que encontramos en sus sistemas digestivos pueden también combinar H
2 (hidrógeno) y CO
2 para producir metano en los sistemas de las fumarolas hidrotermales.
[20] De hecho, muchos metanógenos están adaptados a temperaturas muy altas, de más de 100 grados, y se hallan asociados a las fumarolas hidrotermales modernas.
Incluso sin oxígeno, estos metanógenos crecerían y se multiplicarían, y algunos morirían. La población alcanzaría una especie de estado estacionario, donde el crecimiento compensa a la muerte, y los organismos muertos servirían de comida para otros microbios. En nuestros días, las bacterias fermentadoras representan un papel principal en la descomposición de las sustancias orgánicas producidas por los organismos. Obtienen energía y crecen a través del proceso de fermentación, por el que generan moléculas orgánicas simples que otros microbios pueden usar. De hecho, los metanógenos de un tipo diferente (heterótrofos)
[21] pueden usar esos compuestos orgánicos simples y producir metano y CO
2. Por lo tanto, vemos la posibilidad de un ecosistema basado en el metano, con los metanógenos autótrofos como productores primarios, y una serie de bacterias fermentadoras y metanógenos heterótrofos como consumidores.
Esto, según creo, es una visión realista de los principales jugadores microbianos que poblaron los ecosistemas primitivos en las fumarolas hidrotermales de las profundidades oceánicas antes del advenimiento de los organismos productores de oxígeno. Otras poblaciones de microbios, sin embargo, pudieron haber estado también presentes, aumentando la diversidad del ecosistema. Por ejemplo, si el sulfato se encontrara en el agua marina, aunque fuera en pequeñas concentraciones (veremos mucho más sobre el sulfato en capítulos posteriores), podría sostenerse un proceso conocido como «reducción del sulfato». En este proceso, las bacterias reductoras de sulfato obtienen energía y crecen a través de la reacción del sulfato con la materia orgánica o el H
2; se produce entonces sulfuro y CO
2.
[22] Estos antiguos reductores de sulfato, entonces, podrían haber vivido de la materia orgánica de otros microbios muertos, o del H
2 que emanaba de las fumarolas hidrotermales. Parece probable, por tanto, que los ecosistemas hidrotermales albergaran una población bastante diversa de microbios, pero el H
2 habría sido la fuente primaria de energía más probable.
[23]Los sistemas hidrotermales de las profundidades marinas, sin embargo, no serían los únicos lugares donde se hallaría la vida antes del oxígeno. La joven biosfera, de hecho, probablemente presenció una acción considerable en tierra firme y en las zonas altas del océano global. Hay muchas razones para sostener esto, pero empezaremos por el H
2S (sulfuro de hidrógeno) que revestía un interés tan escaso para la vida en el océano profundo, oscuro y carente de oxígeno. En tierra firme, y a la luz del día, el H
2S se vuelve de repente muy interesante. Cuando estuviera disponible, el H
2S debió ser muy útil para un grupo de organismos fotosintéticos que evolucionaron mucho antes que los mejor conocidos productores de oxígeno. Puesto que estos fotosintetizadores no producen oxígeno, se los conoce como fotótrofos anoxigénicos.
[24] Hablaré mucho de su evolución en el siguiente capítulo, pero aquí introduciré al lector en su ecología. A decir verdad, no son nada raros.
Siempre he soñado con vivir junto al mar y, en un acto de despreocupada indulgencia, mi familia estuvo de acuerdo en comprar una casita en la isla danesa de Bornholm, donde veraneamos todos los años. En la parte suroriental de la isla hay una serie preciosa de playas blancas cuya arena es tan fina y regular que antiguamente se utilizaba para hacer relojes de arena. Precisamente porque la arena es tan fina, se humedece con facilidad y permanece húmeda mucho tiempo, y si arañas con el pie una zona húmeda, encuentras a menudo una serie de capas verdes, rojas y negras muy interesante y reproducible (lámina 2). Si acercas la nariz, puedes sentir el ligero olor del sulfuro de hidrógeno. El sulfuro proviene de la reducción del sulfato, un proceso que ya hemos encontrado anteriormente, y el color negro es producto de la reacción entre el sulfuro y pequeñas cantidades de minerales de hierro en la arena. La capa verde toma su color de las cianobacterias productoras de oxígeno que exploraremos en detalle en los próximos capítulos. Lo más importante para nuestra presente discusión es la banda roja, que recibe su color de los organismos fotosintéticos anoxigénicos. Estos organismos utilizan la energía del Sol para convertir sulfuro en sulfato, y en el proceso generan biomasa celular a partir de CO2.
Aunque la arena de Bornholm proporciona un buen entorno para las poblaciones fototróficas anoxigénicas que usan sulfuro, estas arenas aportan sólo una pobre analogía de la Tierra antigua. Esto se debe a que el sulfuro utilizado por esta población de fotótrofos anoxigénicos se obtiene, en último término, de la descomposición del material orgánico producido por las cianobacterias que pueblan las capas altas de la arena. Si quitáramos las cianobacterias nos quedaríamos sin sulfuro para que lo usaran los fotótrofos anoxigénicos. Un entorno más análogo a la Tierra joven sería el tipo de fuentes termales que se encuentran en el parque nacional de Yellowstone, en Islandia o en la Isla Norte de Nueva Zelanda.
Visité el Parque Yellowstone de niño. Me maravillaron los manantiales de agua caliente y me quedé hipnotizado por el «Old Faithful», pero mi atención permaneció absorbida sobre todo por el avistamiento de osos desde la seguridad de nuestro coche. También recuerdo los colores, sin embargo, los bellos marrones, naranjas, rojos y verdes propagándose como pinturas abstractas desde las gargantas sin fondo aparente de las fuentes hidrotermales. Sólo después de algunos años aprendí que esos colores se formaban por alfombras de bacterias, muchas de las cuales contienen grandes poblaciones de fotótrofos anoxigénicos que oxidan el sulfuro hidrotermal que emana de los manantiales. Esto, en mi opinión, sí es un análogo aceptable de lo que podríamos haber encontrado en la Tierra antigua. Como en el caso de las fumarolas, podemos imaginar el desarrollo de ecosistemas complejos. Los fotótropos anoxigénicos oxidantes de sulfuro producirían sulfato, que sería usado por bacterias reductoras de sulfato para oxidar la materia orgánica producida por los fotótropos. Durante la reducción del sulfato, el sulfato es reducido a sulfuro, lo que recicla el sulfuro para que lo usen de nuevo los fotótropos anoxigénicos. Como en los ecosistemas de la fumarolas hidrotermales, varias bacterias fermentadoras ayudarían a descomponer la materia orgánica y a generar así comida para las bacterias reductoras de sulfato. Este tipo de ecosistema se conoce como sulfuretum, un término introducido por Laurens Baas Becking en 1925 (Baas Becking iniciaría más tarde el campo de la geobiología con su libro de 1934 Geobiologie) (figura 2.1). Un ecosistema así también recicla la materia de una forma directamente análoga a lo que ocurre en nuestra moderna biosfera oxigenada. Basta sustituir el sulfuro por agua, y el sulfato por oxígeno.
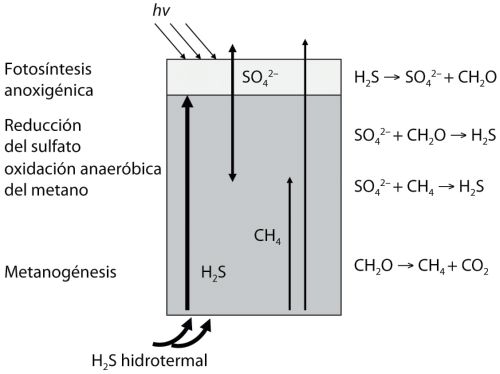
FIGURA 1.2 Posible funcionamiento de un primitivo ecosistema microbiano basado en el azufre, con la probable influencia adicional del reciclado microbiano del metano. El reciclado del metano ocurrirá si parte del sulfato producido durante la fotosíntesis anoxigénica se pierde del sistema, por ejemplo por vertido a un río. Se muestra también el proceso de oxidación anaeróbica del metano, u oxidación del metano con sulfato; es posible que sea significativa, pero no se discute en el texto. Redibujado a partir de Canfield y colaboradores (2006). CH2O indica compuestos orgánicos.
Si parte del sulfato producido por los fotótrofos anoxigénicos fuera retirado con el flujo de agua hidrotermal, quedaría insuficiente sulfato para descomponer toda la biomasa muerta mediante las bacterias reductoras de sulfato. Esta deficiencia permitiría que una comunidad de bacterias productoras de metano se desarrollara y descompusiera el resto del material orgánico, añadiendo incluso más complejidad a nuestro ecosistema alimentado por sulfuro. Si pudiéramos haber visitado esos ecosistemas ancestrales, nos habrían maravillado las coloridas alfombras microbianas, de algún modo análogas a las que encontramos hoy. La vida parecería copiosa en esas áreas hidrotermales. Sin embargo, dada la escasez general de tales áreas a una escala global, los ecosistemas basados en sulfuro probablemente habrían contribuido sólo un poco a la actividad total de la biosfera.
Para encontrar grandes jugadores en la Tierra antes del oxígeno, miremos al cielo y reparemos en que los volcanes primitivos habrían escupido gases como H
2, SO
2 (dióxido de azufre), CO
2 y H
2S a la atmósfera. Como en los sistemas hidrotermales de las profundidades oceánicas, las poblaciones productoras de metano habrían sido sustentadas por el H
2 y el CO
2 volcánicos. Cabe imaginar a metanógenos viviendo en suelos saturados en tierra firme, en lagos y en el mar, a base de combinar CO
2 y gas hidrógeno. De manera más importante, sin embargo, muchas bacterias fotótrofas anoxigénicas pueden convertir el gas hidrógeno (H
2) en agua. Acoplan esta reacción a la generación de biomasa celular a partir de CO
2.
[25] Estos antiguos fotótrofos que utilizan hidrógeno habrían poblado los antiguos lagos, estanques y la superficie del océano; de hecho, habrían vivido en cualquier entorno acuoso iluminado por el Sol en el que el H
2 y el CO
2 de la atmósfera se pudiera disolver. ¿Cuán productivo habría sido ese metabolismo ancestral? Minik Rosing, Christian Bjerrum y yo desarrollamos un modelo concebido originalmente por Jim Kasting y su grupo en Penn State (ya encontramos a Jim en el último capítulo), y estimamos que los fotótrofos anoxigénicos que usaran gas hidrógeno podrían haber producido biomasa a una velocidad máxima de unos 3×10
13 moles de carbono orgánico al año (equivalentes a 3,6×10
14 gramos de carbono al año). Esto parece un gran número, pero sigue siendo cien veces menos que las tasas de producción de carbón orgánico en la biosfera actual, sustentada por la fotosíntesis oxigénica.
Para averiguar cuál puede haber sido el mayor jugador en la biosfera ancestral antes del oxígeno, hagamos una inmersión imaginaria con Alvin a las profundidades de los océanos primitivos. Mientras descendemos, enfoquemos nuestra atención a la profundidad exacta en que la luz solar se disipa en la oscuridad. Aquí podríamos muy bien observar una densa población de bacterias y, curiosamente, una gran abundancia de minerales de óxido de hierro, que se pueden ver básicamente como óxido. Nuestros sensores químicos detectarían también una acumulación de hierro ferroso (Fe2+) disuelto en las aguas profundas, por debajo del lugar donde encontramos las partículas de óxido. ¿Qué ocurre aquí?
Oiremos mucho más sobre el hierro en capítulos posteriores, pero básicamente el hierro ferroso es la forma del hierro que persiste en ausencia de oxígeno, y que se disuelve sin problemas en el agua. Por tanto, sin oxígeno en la atmósfera, el hierro ferroso que proviene de las fumarolas hidrotermales se debió acumular en las profundidades oceánicas ancestrales. De hecho, tenemos evidencias de esto en la acumulación masiva de un tipo peculiar de roca sedimentaria conocida como formaciones de hierro bandeado (BIF en sus siglas inglesas, lámina 3). Estas formaciones son especialmente abundantes en el registro de rocas antes de 2500 millones de años atrás (aprenderemos mucho más sobre las BIF en capítulos posteriores), y nuestros mejores modelos sostienen que esas BIF se formaron a partir del hierro ferroso disuelto en el agua del mar. Pero, centrándonos de nuevo en la capa de partículas, ¿cuál es la fuente de todas esas bacterias, y de todo ese óxido?
El microbiólogo Fritz Widdel, del Instituto Max Planck de Microbiología Marina en Bremen, Alemania, es la viva imagen de la paciencia. De hecho, su paciencia le ha permitido hacer muchos descubrimientos fundamentales en nuestra comprensión del metabolismo microbiano. En uno de esos avances, Fritz y sus estudiantes recogieron barro de un canal de desagüe cercano al Instituto Max Planck,
[26] y lo incubaron bajo la luz añadiendo hierro ferroso. Esperaron, esperaron, esperaron y, finalmente, pasados unos cuantos meses, vieron una población de bacterias purpúreas creciendo en el barro. Las transfirieron a un medio de cultivo y… siguieron esperando. Y entonces por fin, tras todas esas esperas, aislaron una población de bacterias fotótrofas anoxigénicas (lámina 4) capaces de crecer a partir del hierro ferroso y formando, esencialmente, óxido en el proceso. La gente había supuesto desde hacía tiempo que una población semejante debía existir en la naturaleza, pero nadie tenía ni la paciencia de Fritz ni su talento para aislarlas. Sin embargo, ¿habría sido una población semejante de importancia en los océanos primitivos?
Como investigadores del pasado distante, intentamos identificar los entornos modernos que emulan, tan de cerca como sea posible, los ambientes ancestrales que tratamos de entender. ¿Dónde, sin embargo, podríamos encontrar un análogo a un océano rico en hierro de 3000 a 4000 millones de años? Mientras yo estaba reflexionando sobre esto, un estudiante de doctorado canadiense, Sean Crowe, estaba visitando nuestro laboratorio y nos explicó que estaba trabajando en el lago Matano de Sulawesi, Indonesia. Y este resultó ser exactamente el entorno que buscábamos. El lago es profundo, con casi 600 metros, claro y estable. De forma crucial, acumula unas concentraciones de hierro ferroso en las aguas cercanas al fondo. Sean estaba planeando un nuevo viaje a Matano e invitó a mi estudiante de doctorado CarriAyne Jones a ir con él. Sean y CarriAyne aprendieron mucho sobre el lago, pero lo más importante fue que descubrieron una población fotótrofa anoxigénica justo donde el hierro, procedente de las aguas inferiores, se oxida al encontrarse con la pálida luz que llega de arriba. Aunque estos fotótrofos han evadido el aislamiento, Sean y CarriAyne concluyeron, tras una variedad de consideraciones, que lo más probable es que oxiden el hierro ferroso a óxido en el lago. El lago Matano está muy lejos del desagüe de Fritz Widdel, pero ambos entornos apuntan a la posibilidad de que bacterias fotótrofas anoxigénicas oxidantes de hierro pudieran haber hecho una contribución importante a la productividad biológica de la Tierra primitiva.
Imaginamos que estos organismos oxidantes de hierro habrían sido socios en ecosistemas que también contaban con bacterias fermentadoras y las denominadas bacterias reductoras de hierro. Los reductores de hierro son un grupo bien conocido de microbios que crecen reduciendo el óxido férrico de vuelta a hierro ferroso, oxidando materia orgánica o H
2 en el proceso. En los océanos primitivos, habrían hecho el mismo trabajo recombinando los productos de la fotosíntesis, los óxidos de hierro y la biomasa celular, oxidando la biomasa celular para dar CO
2, y reduciendo los óxidos de hierro para volver a dar hierro ferroso disuelto en el agua (figura 2.2). Este ecosistema reciclaría eficazmente el hierro, y la actividad de los fotótrofos vendría controlada en último término por la disponibilidad de hierro ferroso y nutrientes. Minik, Christian y yo también intentamos hacer un modelo para estimar el nivel de actividad de esta primitiva población fotótrofa. Nuestros cálculos resultaron muy imprecisos y cargados de suposiciones e incertidumbres,
[27] pero, reconociendo esas limitaciones, calculamos que una biosfera basada en el hierro podría haber tenido, quizá, el 10% de la actividad de la biosfera marina actual. No tan mezquino.
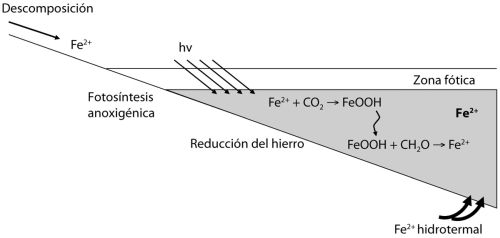
FIGURA 2.2 Posible estructura de un ecosistema basado en el hierro en los océanos. Los detalles se explican en el texto. Redibujado a partir de Canfield y colaboradores (2006).
Resumamos. Parece probable que, antes de la evolución de los organismos productores de oxígeno, existieran en la Tierra numerosos ecosistemas interesantes y dinámicos en entornos que abarcarían desde las profundidades oceánicas hasta la superficie del mar y los sistemas hidrotermales basados en tierra firme, así como en los lagos y en los suelos. En algunos casos estos ecosistemas habrían sido algo diminutos, y en otros, muy obvios a simple vista. El ecosistema más activo pudo haber estado basado en la oxidación del hierro ferroso disuelto en los océanos. Parece probable, sin embargo, que la biosfera ancestral fuera mucho menos activa que la actual, que está conducida por organismos productores de oxígeno.
¿Podemos hallar alguna evidencia de esos ecosistemas antiguos en el registro geológico? Esto dista de ser seguro. Una dificultad, que exploraremos en más detalle en el capítulo 7, es que realmente no sabemos cuándo evolucionaron las cianobacterias productoras de oxígeno. Por tanto, no estamos seguros de cuánto debemos remontarnos en el tiempo para tener confianza en que las rocas que exploramos se depositaron en un mundo carente de organismos productores de oxígeno. E incluso descontando esto, no hay muchas de esas rocas antiguas por ahí, y las que podemos encontrar no están en buena forma. Como exploraremos en el capítulo 6, la misma tectónica de placas que hace del mundo un lugar tan habitable deja un registro geológico fragmentado y comprometido. Los volteos, mezclas y movimientos del planeta promueven la erosión física y química de las rocas antiguas y también estimulan su enterramiento, calentado y deformación. En breve, la mayoría de las rocas que antaño ocuparon la superficie del planeta ya no están allí, y muchas de las que quedan han sufrido calor y una gran deformación, y este problema se vuelve peor cuanto más atrás en el tiempo miremos.
Aun así, a pesar de las injurias de la edad, hay algunas pistas sobre los tipos de vida microbiana que poblaron los ecosistemas muy antiguos. De hecho, un lugar muy revelador para investigar son las rocas de casi 3500 millones de años de North Pole en Australia.
[28] Estas rocas están notablemente bien conservadas para su edad, y han sido estudiadas de forma intensa por mi colega australiano Roger Buick, ahora en la Universidad de Washington. Roger pinta una imagen de un terreno volcánico activo al borde del océano. Imagine el lector unas lagunas marinas semiaisladas que mojan a los vertidos basálticos y acumulan sedimentos en un entorno con altas concentraciones locales de sulfato en el agua. La fuente de sulfato era probablemente, en último término, los gases sulfurosos que desprendían los volcanes de la zona. Los minerales de sulfato, algunos de los cuales se depositaron originalmente en forma de yeso (todos son ahora barita, BaSO
4), están asociados a piritas de grano fino y forman un componente importante de los sedimentos. Mi entonces
posdoc (investigador posdoctoral) Yanan Shen, junto a Roger Buick y yo mismo, examinamos la composición isotópica del azufre en los sulfatos y piritas de esas rocas. Diré más sobre lo que esos isótopos nos revelan en los capítulos 7 y 9, pero nuestros resultados indicaron que el sulfuro de las piritas se formó por reducción microbiana de sulfatos. Nos sentimos orgullosos de ese resultado, porque documentaba tanto la evolución temprana de las bacterias reductoras de sulfato como el metabolismo microbiano específico más antiguo que se conocía en el registro geológico.
Como cabía esperar, este hallazgo fue sometido a un escrutinio intenso, con la discusión centrada en si el sulfuro pudo formarse por un proceso termoquímico sin implicación de organismo alguno. Esto puede suceder si se calienta el sulfato junto a materia orgánica a una temperatura lo bastante alta. Yanan Shen ha revisitado desde entonces esas rocas con una estrategia de isótopos de azufre más sofisticada y ha generado nuevos resultados que son coherentes con nuestro hallazgo anterior.
Hay más aún. Las rocas basálticas que yacen justo debajo de los sedimentos explorados por Yanan, Roger y yo aparecen entrecruzados con varios canales ricos en sílice, y estos contienen diminutas inclusiones de fluidos y gases. Como los canales se formaron al mismo tiempo que los sedimentos, estas inclusiones podrían, en principio, contener más pistas sobre los microbios que vivieron hace 3500 millones de años. De hecho, Yuichiro Ueno, del Instituto de Tecnología de Tokio, y sus colegas han examinado los gases presentes en esas inclusiones y han hallado que muchas contienen bastante metano. Han dividido las inclusiones entre aquellas que parecen primarias, que datan de la misma época en que se formaron los canales, y otras que parecen secundarias, formadas después del emplazamiento original de los canales. Luego han medido la composición isotópica del metano, puesto que el metano biogénico da una señal isotópica distinta. El resultado es que las inclusiones primarias contienen una composición isotópica del metano (no se preocupe por los detalles) coherente con un origen biogénico a través de la metanogénesis microbiana. En contraste, las inclusiones secundarias presentan composiciones isotópicas más coherentes con fuentes no biogénicas. Esto constituye una bonita pieza de investigación detectivesca en versión geoquímica, y muestra que los organismos productores de metano también eran parte del ancestral ecosistema microbiano de North Pole, Australia.
En conclusión, las evidencias geológicas implican que muchos de los procesos que hemos imaginado fueron en verdad parte de la biosfera ancestral que funcionaba hace 3500 millones de años. Estos procesos incluyen la metanogénesis, la reducción del sulfato y la descomposición de la biomasa orgánica muerta, que fue probablemente asistida por una multitud de diferentes bacterias fermentadoras. Por desgracia, los indicadores de la fotosíntesis anoxigénica en rocas de tal antigüedad no son demasiado robustos, de modo que el registro geológico resulta más bien silencioso acerca de la antigüedad de este proceso. Con todo, exploraremos otras formas de analizar la antigüedad de la fotosíntesis anoxigénica en el siguiente capítulo.
Puede haber, sin embargo, otra manera de investigar la historia ancestral de la evolución microbiana que no depende del registro geológico. La premisa es simple. Todos los organismos de la Tierra contienen un registro de su historia evolutiva en su ADN. Esto se debe a que el ADN de cualquier organismo, incluidos nosotros, es el producto de todos los cambios que han ocurrido en su linaje antes del momento presente. La historia grabada en el ADN, sin embargo, es complicada. Está influenciada por numerosos factores, como la duplicación de genes, el nacimiento de nuevos genes, la pérdida de genes, la transferencia de genes entre distintos organismos y todas las mutaciones acumuladas en la secuencia de ADN a través del tiempo. En principio, de todos modos, podemos comparar el ADN de un organismo con el de otro para entender cómo el ADN difiere entre ellos, y si comparamos el suficiente número de organismos diferentes podemos comprender la historia de la evolución del ADN. Eric Alm, del Massachusetts Institute of Technology (MIT) y su estudiante Lawrence David han echado un vistazo particularmente sofisticado a este asunto. Han analizado casi 4000 genes en más de 100 organismos, y han ofrecido así una historia de la evolución genética. Los resultados son fascinantes.
He reproducido algunos de esos resultados en la lámina 5. Lo que vemos es una historia de la importancia de los diferentes tipos metabólicos a lo largo de la historia de la Tierra. Hay muchas suposiciones detrás de la construcción de esa historia evolutiva, y este es uno de los primeros intentos de emplear esta fascinante estrategia. Pero, con todo esto en mente, yo clavo los ojos en estos resultados. Estos datos son, en principio, exactamente lo que estamos buscando. ¿Qué tipo de organismos definieron la biosfera hace 3500 millones de años? ¿Cuándo evolucionó la reducción del sulfato? ¿Y la metanogénesis? Los resultados de la lámina 5 indican que el ciclo del azufre estaba activo muy pronto en la historia de la Tierra. Los metabolismos autótrofos también evolucionaron temprano, mientras que los genes para la utilización del oxígeno se expandieron más recientemente. De acuerdo con este análisis, sin embargo, la metanogénesis es un metabolismo que evolucionó más tarde. ¡Ups! Yo encuentro esto difícil de creer, sobre todo en vista de las evidencias geológicas descritas más arriba, pero, como he dicho, estamos mirando a la aplicación inicial de esta estrategia, no a la última.
Tratemos de poner todo esto junto. Por cualquier cálculo, la Tierra disfrutaba de una biosfera activa y diversa mucho antes de la evolución de las cianobacterias productoras de oxígeno. Aquella biosfera estaba alimentada, principalmente, por compuestos químicos liberados por el volcanismo, lo que subraya de nuevo la importancia de la tectónica de placas para dar forma a la vida en nuestro planeta. El registro geológico viene en apoyo de una primitiva biosfera diversa, al igual que las nuevas estrategias moleculares dirigidas a descifrar la historia evolutiva a partir de secuencias de ADN. Parece probable, sin embargo, que esta biosfera temprana fuera mucho menos activa que la que disfrutamos en el presente. En los próximos capítulos empezaremos a examinar los pasos que condujeron a la evolución de los organismos productores de oxígeno y comenzaremos a entender cómo se formó la biosfera actual
Capítulo 3
La evolución de la fotosíntesis oxigénica
La carrera había empezado, pero realmente nadie sabía que había una carrera, al menos no al principio. Corría el año 1771 (o tal vez 1772), y el farmacéutico sueco Carl Wilhelm Scheele estaba verdaderamente muy ocupado. Habiendo sólo empezado como ayudante de laboratorio del químico Torbern Bergman, de Upsala, Scheele estaba concentrado en esclarecer los misterios del aire. Su motivación real, sin embargo, tenía más de chiquillada que todo eso; lo que realmente quería entender era la naturaleza del fuego. Tal y como Scheele admitió, «uno no puede formarse ningún juicio verdadero sobre el fenómeno que presenta el fuego sin un conocimiento del aire».
[29]En aquel momento de la historia de la ciencia, el aire era una sustancia verdaderamente extraordinaria y misteriosa. Podemos apreciarlo viendo cómo el propio Scheele resumió sus propiedades: «El aire es esa sustancia fluida e invisible que respiramos continuamente, que rodea toda la superficie de la Tierra, es muy elástico y tiene peso». Además de estos hechos exiguos, se había demostrado que el aire contenía dióxido de carbono, pero aparte de eso se sabía francamente poco. Parte del problema era que el concepto de los elementos químicos apenas estaba emergiendo, y los que constituyen los componentes del aire no habían sido descubiertos todavía. Otro problema era que la naturaleza del aire no podía desacoplarse de la entonces popular teoría del flogisto. Se creía que el flogisto era una sustancia incolora sin masa que las sustancias inflamables liberaban al quemarse. De este modo, el aire se volvía flogistado cuando los objetos ardían, y las sustancias combustibles no podían seguir ardiendo cuando el suficiente flogisto se había acumulado en el aire. Esta explicación, sin embargo, era probablemente insuficiente para Scheele, que se dio cuenta de que la verdadera clave para entender el proceso de la combustión era comprender la naturaleza del aire.
Se puede seguir el proceso de pensamiento de Scheele en su libro
Tratado químico del aire y el fuego, publicado en 1777 y ahora traducido al inglés. En experimentos iniciales, Scheele quemó, entre otras cosas, una variedad de compuestos azufrados para confirmar una propiedad del aire que ya era conocida. Esto es: «Las sustancias sujetas a putrefacción o a destrucción por el fuego disminuyen, y al mismo tiempo consumen, una parte del aire». De lo que Scheele concluyó que «el aire debe estar compuesto de fluidos elásticos de dos clases». Pero ¿cuál era la naturaleza de esas dos clases de fluidos? Los experimentos continuaron. El verdadero descubrimiento llegó cuando Scheele destiló nitrato potásico en una mezcla de ácidos nítrico y sulfúrico (¡feo asunto en verdad!) y recogió gases casi al final de la destilación, cuando «se producen unos vapores rojos como la sangre».
[30] Y, para su sorpresa, si una vela se introduce en este gas, «no sólo continuará ardiendo, sino que lo hará con una luz mucho más brillante que en el aire ordinario». Luego generó el mismo producto de otras varias formas y lo llamó «fuego-aire». Entonces estimó que ese producto compone alrededor de un tercio de nuestra atmósfera.
[31] Lo que Scheele había encontrado, por supuesto, era oxígeno. Mediante una serie de ingeniosos experimentos adicionales con plantas, ratas e insectos, llegó a conjeturar que el «fuego-aire» es reemplazado por (o convertido en, según él) dióxido de carbono mediante la respiración, y que el «fuego-aire» es absorbido por los pulmones de los animales y transportado por la sangre a todo el cuerpo.
Scheele descubrió que el aire consiste en oxígeno, dióxido de carbono y una gran porción de una sustancia inerte; llamó a esta última «aire viciado», y es lo que ahora conocemos como gas nitrógeno. En cuanto al «fuego-aire», Scheele reflexionó: «Me inclino a creer que el fuego-aire consiste en una sutil sustancia ácida unida con flogisto». Esta conclusión, sin embargo, debió parecerle deficiente, puesto que en septiembre de 1774 envió una carta al químico francés de fama mundial Antoine Lavoisier explicando sus experimentos y pidiéndole consejo.
Irónicamente, al mismo tiempo y sin conocimiento de Scheele, el inglés Joseph Priestley estaba en París discutiendo sus propios experimentos sobre la producción de oxígeno con Lavoisier en persona.
[32] Lavoisier debió de escuchar atentamente, porque poco después produjo oxígeno por su cuenta; sin vínculos con los adornos de la teoría del flogisto, reconoció el oxígeno correctamente como un elemento y le dio el nombre que usamos ahora (que significa «productor de ácido»). Él también, como Scheele, exploró el papel del oxígeno en la respiración y, de forma más precisa que Scheele, estimó que el oxígeno da cuenta del 25% de la atmósfera de la Tierra.
Muchos se han quejado de que Lavoisier dio un escaso crédito a Priestley y ningún reconocimiento a Scheele en sus propios escritos sobre el oxígeno. En verdad, Lavoisier aseguró que nunca había recibido la carta de Scheele, aunque la misiva apareció entre los efectos de la mujer de Lavoisier en la década de 1890. ¿Recibió Lavoisier la carta y la sepultó entre las cosas de su mujer para poder reclamar el crédito de haber descubierto el oxígeno? ¿O recibió la esposa la carta y la escondió de manera que Lavoisier pudiera recibir el crédito sin el conocimiento de un competidor?
[33] Nunca lo sabremos, pero está claro que, como ocurre a menudo en ciencia, nuestra capacidad para explicar el mundo bajo el paradigma existente se vuelve tan inadecuada que una nueva forma de pensar se hace necesaria. Claramente, el tiempo estaba maduro al empezar la década de 1770 para el descubrimiento del oxígeno.
Continuemos con nuestra breve digresión histórica. Aunque Scheele experimentó extensivamente con plantas, nunca descubrió el proceso de la fotosíntesis. Priestley sí lo hizo, en cambio, y en 1771, incluso antes de descubrir el oxígeno, ya había encontrado que las plantas aportaban una sustancia que podía mantener vivo a un ratón y sostener la combustión de una vela. La mayoría de los autores, sin embargo, acreditan al médico holandés Jan Ingenhousz por entender a fondo cómo funciona la fotosíntesis. Para 1779, Ingenhousz fue capaz de resumir su descubrimiento, y en unos términos asombrosamente modernos: «Parecerá quizá probable que uno de los grandes laboratorios de la naturaleza para limpiar y purificar el aire de nuestra atmósfera está situado en la sustancia de las hojas, y puesto en acción por la influencia de la luz».
[34] Como se puede ver, hacia el final del siglo XVIII había ya un entendimiento razonablemente bueno de la principal composición química de la atmósfera, incluido el origen del oxígeno y su papel en la respiración.
En lo que queda de este capítulo seguiremos explorando la historia, pero de otra clase. Nuestro objetivo será comprender la evolución de la producción fotosintética de oxígeno en la Tierra. Espero que podamos estar de acuerdo en que este evento fue uno de los principales logros transformadores en la historia de la vida. Sin fotosíntesis oxigénica no habría oxígeno en la atmósfera; tampoco habría plantas, ni animales ni nadie para contar esta historia.
FIGURA 3.1 Cosecha de la luz en un organismo fotosintético. La figura muestra la relación entre el complejo antena y el centro (o centros) de reacción, cada uno de los cuales contiene pigmentos captadores de luz. Redibujado a partir de Canfield y colaboradores (2005).
Como una innovación biológica, ¿de dónde vino la producción de oxígeno? Para desentrañar este problema, debemos empezar por examinar cómo funciona la producción de oxígeno por fotosíntesis. Todo empieza con la luz, y en la figura 3.1 he reproducido a grandes rasgos cómo la luz (hv) es capturada durante el proceso fotosintético. Este cuadro general se aplica a todos los tipos de fotosíntesis, incluida la fotosíntesis por cianobacterias, los primeros productores de oxígeno (como exploraremos en el capítulo 4). También se aplica a las plantas, e incluso a los fotótrofos anoxigénicos que vimos en el primer capítulo. Para recoger la luz, todos los fotótrofos usan un «complejo antena». Está hecho de una variedad de pigmentos que capturan la luz (figura 3.1). Una vez capturada, la energía de la luz se transfiere a los centros de reacción. Estos constituyen la terminal de negocios del proceso donde, en el caso de los fotótrofos oxigénicos, se produce el oxígeno. La naturaleza ha hecho las cosas complicadas y, en los productores de oxígeno, hay en realidad dos centros de reacción acoplados: uno se conoce como fotosistema I (PSI) y el otro como fotosistema II (PSII). Se acoplan juntos en lo que se suele llamar esquema Z, una estructura que tiene mucho sentido, como espero que resulte obvio más abajo.
Para entender cómo funciona la fotosíntesis oxigénica, nuestro objetivo simple será seguir a los electrones. El proceso se delinea en la figura 3.2, y empezaremos con PSII. La energía del complejo antena es transferida a una molécula especial de clorofila llamada P680. Esta energía hace que P680 desarrolle un estado excitado llamado P680*, que se vuelve así un fuerte reductor (fuente de electrones). Como un niño gritando para ir al cuarto de baño, P680* se deshace fácilmente de un electrón y se lo pasa a un compuesto químico conocido como feofitina, reduciendo la feofitina en el proceso. Nos ocuparemos de lo que le pasa a la feofitina en un minuto, pero nuestro interés inmediato es la clorofila P680 que acaba de perder un electrón. Este electrón debe ser repuesto, pues de lo contrario el proceso entero se bloqueará.
FIGURA 3.2 Los centros de reacción acoplados en la fotosíntesis oxigénica, el llamado esquema en Z. El truco para comprenderlo es seguir a los electrones, como se explica en el texto. Figura cedida cortésmente por Raymond Cox.
En la notable innovación de la fotosíntesis oxigénica, ese electrón viene del agua. Esto es un problema importante porque, como sabemos por experiencia, el agua es una sustancia estable: no nos preocupa, por ejemplo, que pueda empezar a burbujear oxígeno cuando nos bañamos. La belleza de P680 es que al perder su electrón se vuelve un oxidante muy fuerte (un aceptor de electrones, de hecho el más fuerte conocido en la naturaleza), lo que significa que puede tomar electrones del agua, generando oxígeno como un subproducto. Es verdad que todo esto suena más fácil de lo que es realmente. Conseguir que los electrones fluyan desde el agua hasta P680, con la formación de oxígeno, requiere un ballet bioquímico cuidadosamente orquestado. Esta danza es dirigida por el llamado complejo de evolución del oxígeno (oxygen-evolving complex, OEC), que contiene en su núcleo un racimo cuádruple de Mn (manganeso). Nos ocuparemos de él en breve.
Pero antes de eso volvamos a la feofitina. Cuando obtiene su electrón de P680*, lo hace deprisa (en 3 picosegundos, de hecho; ¡eso es 3 billonésimas de segundo!); esta rapidez es esencial para evitar que el electrón se recombine de nuevo con la P680 oxidada, que, como vimos antes, es un fuerte oxidante y es muy bueno extrayendo electrones. Si lo hiciera, el proceso entero se pararía de forma prematura. En último término, la célula tiene que poner el electrón de la feofitina en un transportador de electrones soluble conocido como NADP(H).
[35] Una vez empaquetado en el NADP(H), el electrón puede usarse para dirigir toda clase de reacciones bioquímicas para la célula. Sin embargo, la feofitina apenas es capaz (no tiene el poder reductor suficiente, en términos químicos) de transferir su electrón al NADP(H), y aun si lo hiciera ahora, la célula tendría muy poco que ganar del proceso fotosintético.
En vez de eso, el electrón se utiliza para hacer trabajo. En lo que sigue, podemos imaginar al electrón deslizándose pendiente abajo, como un
kart en un agradable día de verano. El electrón se desliza de la feofitina a una molécula de quinona y sigue cuesta abajo a través de una serie de proteínas. La célula se aprovecha de este viaje despreocupado y, a medida que el electrón rueda pendiente abajo, se forma ATP.
[36] El ATP es la moneda energética de la célula y, en términos descarnadamente opuestos al romanticismo, se puede defender que el propósito de la vida es hacer ATP. En cualquier caso, cuando el electrón llega a la base de la montaña, no queda mucha energía en la proteína que lo transporta. Ya no hay forma de que haga NADP(H) ni de que haga gran cosa por la célula. Así que el electrón se entrega al PSI (fotosistema I). Aquí hay otra molécula de clorofila conocida como P700, cuya forma oxidante permanece en espera de este electrón. Cuando P700 se une al electrón, energía procedente de del complejo antena (
hv) impulsa a P700 a un estado excitado y reducido, P700*, que de hecho es un reductor más poderoso que la P680* excitada que acabamos de discutir. El viaje cuesta abajo del
kart empieza de nuevo, pero como el electrón parte de un nivel más alto (un estado más reducido), permanece lo bastante reductor como para combinarse fácilmente con el NADP+ para formar NADP(H).
Todavía no hemos acabado. Si lo dejáramos aquí, los electrones empaquetados en el NADP(H) no harían más que acumularse. Esto no podría ocurrir, y de hecho no ocurre. En realidad, los electrones del NADP(H) que, recordemos, provienen en último término del agua, se transfieren al CO
2, produciendo los compuestos orgánicos que se usan para construir las células. Este proceso se conoce como «fijación del carbono» y la incorporación del CO
2 se promueve por una enzima llamada Rubisco.
[37] De hecho, la fijación de carbono promovida por Rubisco es la base virtual de toda la comida que comemos y de casi toda la energía fósil que usamos. Ha sido un largo viaje en verdad, pero al final la célula consigue justo lo que necesita para crecer, y expulsa O
2 en el proceso como un producto de desecho.
Con esta breve descripción como telón de fondo, examinaremos la evolución de los organismos productores de oxígeno mediante la consideración de la evolución, y el ensamblaje, de sus partes constituyentes básicas. Nos centraremos en las siguientes cuestiones clave: (1) ¿Cuál es la historia evolutiva de la clorofila? (2) ¿Cuáles son las historias evolutivas de PSI y PSII? (3) ¿Cuál es el origen del complejo de evolución del oxígeno en PSII? Y finalmente, (4) ¿Cuál es la historia evolutiva de Rubisco? Analizando estas cuestiones, buscaremos comprender el complejo camino que conduce a la evolución de la fotosíntesis oxigénica en la Tierra.
Como ya sugerimos en el último capítulo, la fotosíntesis oxigénica probablemente no fue el primer tipo de fotosíntesis; ese honor corresponde a las bacterias fotosintéticas anoxigénicas. No dedicaré mucho tiempo a explorar la evolución de estos organismos pioneros de la fotosíntesis, un asunto del que no se sabe gran cosa. Como veremos, sin embargo, la evolución de la fotosíntesis oxigénica tiene más sentido cuando se examina a la luz de la fotótrofos anoxigénicos como precursores. Por tanto, volveremos la vista a los fotótrofos anoxigénicos más de una vez para buscar pistas sobre el origen de la fotosíntesis oxigénica.
Empecemos por la clorofila. Recuerdo bien la frustración de mi madre cuando yo intentaba colarme por la puerta trasera con aquellas manchas verdes brillantes en mis vaqueros nuevos o, peor aún, en mi camisa blanca de los domingos. Aparte de desafiar a las madres (y los padres) a mantener la ropa de los niños en un estado razonable de limpieza, la clorofila que produce esas manchas verdes, como vimos antes, también cumple varias funciones diferentes en un organismo productor de oxígeno; es parte integral del complejo antena y un componente esencial de los dos fotosistemas. La razón principal de su importancia para la fotosíntesis oxigénica, sin embargo, es que produce la forma altamente oxidante de P680 en PSII, que puede extraer electrones del agua. Esta es la gran innovación de la fotosíntesis oxigénica.
Así que, ¿de dónde viene la clorofila? Resulta que la clorofila no es en absoluto una molécula particularmente rara. Tiene relación estructural y química con otras moléculas muy comunes (las llamadas porfirinas) utilizadas ampliamente en todo tipo de enzimas celulares, incluido el grupo hemo de la hemoglobina. La clorofila también está relacionada muy estrechamente con las bacterioclorofilas usadas en los organismos fotosintéticos anoxigénicos. En realidad, las rutas sintéticas de la clorofila a y las diversas bacterioclorofilas son muy similares y divergen sólo en los pasos puramente finales. Por tanto, hay una pequeña distancia bioquímica entre la clorofila, la bacterioclorofila e incluso las porfirinas comunes que utilizan nuestras células. La cuestión, sin embargo, es cuál vino primero.
La mayoría de los bioquímicos estaría de acuerdo en que las profirinas, como clase general de moléculas, evolucionaron antes que la clorofila y la bacterioclorofila. Estas moléculas pioneras de porfirina habrían ayudado a promover la bioquímica de la vida más antigua de la Tierra. Si consideramos ahora los pigmentos fotosintéticos clorofila y bacterioclorofila, podemos suponer que la clorofila evolucionó primero, al menos si basamos esta afirmación en las rutas de formación de estas moléculas. Esta idea fue presentada por Sam Granick en 1965 (se conoce como la hipótesis de Granick) y se basa en la idea de que la clorofila a se forma en un solo paso a partir de su precursor inmediato, una molécula llamada clorofílido a, mientras que formar la bacterioclorofila a desde la misma molécula precursora supone varios pasos. Por tanto, la clorofila a es más fácil de hacer.
Pero podemos mirar a este problema desde otro ángulo. En nuestra era genómica, las historias evolutivas pueden reconstruirse directamente a partir de las secuencias de ADN de los organismos. Vimos una elegante aplicación de esto al final del último capítulo, donde David y Alm exploraron la historia evolutiva de los genes que dirigen una amplia variedad de distintos metabolismos microbianos. Usando una estrategia relacionada, Jin Xiong y Carl Bauer, de la Universidad de Indiana, exploraron la historia evolutiva de los organismos fotosintéticos oxigénicos y anoxigénicos. Como señalé antes, la clorofila y la bacterioclorofila se forman por la misma ruta bioquímica excepto por los pasos finales de la biosíntesis. Por tanto, se pueden identificar genes en los organismos fotótrofos tanto oxigénicos como anoxigénicos que dirigen exactamente el mismo proceso en la síntesis de ambas moléculas, la clorofila y la bacterioclorofila. Xiong y Bauer determinaron las secuencias de ADN de varios de estos genes, y al comparar las secuencias de los mismos genes, las de las bacterias fotótrofas anoxigénicas siempre parecían más antiguas que las de los fotótrofos oxigénicos. Esto es una buena evidencia de que la biosíntesis de clorofila es más moderna que la biosíntesis de la bacterioclorofila, lo que indica que la bacterioclorofila vino primero. Por tanto, el hecho de que la clorofila tenga una ruta biosintética final algo más simple debe compensarse contra otras evidencias de que la bacterioclorofila, de hecho, se originó antes.
Así, nuestra idea de que la fotosíntesis anoxigénica precedió a la fotosíntesis oxigénica parece sujetarse bastante bien, como también lo hace la idea de que la biosíntesis de clorofila emergió después, quizá junto a la evolución de la fotosíntesis oxigénica. Pero ¿por qué? ¿Por qué los organismos usaron primero bacterioclorofila, el producto biosintético más complejo, en lugar de empezar directamente con la clorofila? Tal vez la ruta de la bacterioclorofila evolucionó primero por azar y, puesto que funcionaba, no hubo presión evolutiva para incorporar un nuevo sistema de pigmentos. O tal vez, como han propuesto Martin Hohmann-Marriott, de la Universidad de Otago, y Bob Blankenship, de la Universidad de Washington (de quien volveremos a hablar más abajo), las primitivas enzimas que formaban la bacterioclorofila podrían haber dirigido múltiples pasos en el proceso de síntesis, puenteando esencialmente el paso de la clorofila, o produciendo clorofila sólo como un producto menor. Sólo más tarde, cuando evolucionaron los genes adecuados, la síntesis de clorofila se habría vuelto una ruta sintética principal en los organismos productores de oxígeno. Esto parece posible, pero lo cierto es que aún tenemos mucho que aprender.
Dirijamos ahora nuestra atención a los centros de reacción, PSI y PSII, el corazón del proceso fotosintético oxigénico. Bob Blankenship, de la Universidad de Washington, ha estudiado la fotosíntesis muchos años y, junto a su antiguo estudiante Jason Raymond, han pensado sobre la evolución de la fotosíntesis probablemente más que nadie. Se dice que una imagen vale más que mil palabras, y hace tiempo que Bob halló similitudes funcionales y estructurales entre PSI y PSII, y los centros de reacción utilizados por los fotótrofos anoxigénicos. Tratemos de ver lo que vio Bob con ayuda de la figura 3.3. Al comparar las rutas bioquímicas de PSI y PSII con las de los fotótrofos anoxigénicos, Bob pudo ver grandes parecidos entre PSI y el centro de reacción de «tipo 1» (o tipo FeS) utilizado por algunos fotótrofos anoxigénicos, incluido un grupo conocido como «bacterias verdes del azufre», o cariñosamente GSB (por
green sulfur bacteria).
[38] De forma crítica, tanto en PSI como en el centro de reacción de tipo 1 los electrones son transferidos a través de una serie similar de proteínas después de emerger de las moléculas de clorofila o bactrerioclorofila altamente activadas que describimos antes. Cuando Bob miró a PSII, también apreció muchas similitudes con los centros de reacción de «tipo 2» (o tipo Q) utilizados por otros fotótrofos anoxigénicos. Este centro de reacción de tipo 2 tiene parecidos estructurales con PSII y también transfiere electrones de forma similar. Muchos de los organismos fotótrofos anoxigénicos que usan el centro de reacción de tipo 2 forman parte de un grupo conocido como bacterias púrpuras.
[39] Estos fotótrofos anoxigénicos púrpuras son un grupo muy diverso, pero todos usan el mismo tipo de centro de reacción.
FIGURA 3.3 Comparación de los centros de reacción de los fotótrofos anoxigénicos con los fotosistemas acoplados de los productores de oxígeno. No hay que preocuparse por los detalles, sino percibir las similitudes entre PSII y el centro de reacción de tipo I de las bacterias púrpura; y entre PSI y el centro de reacción de tipo II de las bacterias verdes del azufre. Tomado con modificaciones de Blankenship (2010), reproducido con permiso.
Con ser esto ya muy convincente, Bob y Jason, junto a Sumedha Sadekar, han llevado la comparación de los centros de reacción a otro nivel. En los últimos años, la tecnología ha permitido la determinación de la estructura de grandes proteínas con una resolución tan asombrosa que, en muchos casos, se pueden distinguir los átomos individuales. Con esta resolución, los parecidos y diferencias de estructura entre las diversas proteínas de los centros de reacción pueden determinarse con certeza. La lógica de estas comparaciones es que las proteínas con estructuras similares están más relacionadas evolutivamente. Así, cuando comparamos la estructura de las proteínas de PSI, PSII y los centros de reacción de tipo 1 y 2 (figura 3.4; véase lámina 6 para una versión en color), apreciamos un parecido considerable, sobre todo en las regiones centrales, donde la proteína está embebida en una membrana interior de la célula. Esto es una buena evidencia de que todas las proteínas de los centros de reacción están relacionadas entre sí. Con análisis adicionales de la estructura de estas proteínas, Sadekar, Raymond y Blankenship concluyeron, de forma coherente con la propuesta original de Bob Blankenship, que las proteínas fotosintéticas anoxigénicas de tipo 1 y tipo 2 son los precursores ancestrales de PSI y PSII.
El lector probablemente adivinará adónde va esto. Una explicación lógica de todas estas observaciones es que los centros de reacción PSI y PSII se derivaron de los centros de reacción de tipo 1 y tipo 2 que ya existían en las bacterias fotosintéticas anoxigénicas. De alguna manera, y los detalles no están claros en absoluto, estos dos centros de reacción preexistentes se conectaron para formar los fotosistemas acoplados en las bacterias fotosintéticas oxigénicas. La conexión puede haber ocurrido cuando el centro de reacción de un tipo se transfirió a un organismo que contenía el centro de reacción del otro tipo; o bien, los dos tipos de centros de reacción estaban acoplados ya en un organismo fotosintético anoxigénico original, que después desarrolló la producción de oxígeno fotótrofa. En cualquiera de los dos escenarios, organismos fotótrofos anoxigénicos contuvieron en algún momento ambos tipos de fotosistemas. Si el primer escenario es el correcto, el organismo fotótrofo anoxigénico precursor, con los dos fotosistemas, aparentemente ha desaparecido de la naturaleza (pero véase más abajo). Si el correcto es el segundo escenario, las modernas bacterias púrpuras y GSB han perdido cada una uno de los dos centros de reacción. Bob Blankenship sostiene que por el momento es imposible distinguir entre las dos posibilidades, y tal vez lo sea. Sin embargo, a John Allen, de la Universidad de Londres, le gusta la idea de que los dos centros de reacción existieron una vez juntos en el mismo fotótrofo anoxigénico, quizá derivados de un solo precursor mediante un evento de duplicación genética.
[40] Desde su punto de vista, cada uno de los fotosistemas era útil al organismo, y uno u otro era elegido para funcionar dependiendo de las condiciones ambientales. En algún momento los dos fotosistemas se combinaron. En opinión de Allen, algunos fotótrofos anoxigénicos pudieran muy bien existir hoy con ambos centros de reacción intactos. ¡Sería realmente emocionante hallar un organismo semejante!
FIGURA 3.4 Comparación de las estructuras de las proteínas del centro de reacción fotosintético. Se indica también si la proteína es de un centro de reacción fotosintético anoxigénico de tipo I o tipo II, o si son PSII o PSI de cianobacterias fototróficas oxigénicas. Tomado con ligeras modificaciones de Sadekar y colaboradores (2006), reproducido con permiso. Véase la lámina 6 para una versión en color.
Aunque estamos más cerca de entender la evolución de los organismos fotosintéticos oxigénicos, tenemos aún que explicar cómo empezó a producirse el oxígeno. Para ello tenemos que centrarnos en el complejo de evolución del oxígeno (OEC), que contiene en su núcleo un racimo de 4 átomos de Mn y uno de Ca (calcio). La forma exacta en que funciona es todavía objeto de debate activo, pero es bien sabido que los átomos de Mn son las bestias de carga aquí. Estos átomos actúan básicamente como un condensador biológico. Para generar O2, la forma oxidada de P680 retira 4 electrones del complejo Mn (no directamente del agua). Estos 4 electrones son reemplazados cuando dos moléculas de agua liberan electrones, y de este modo liberan O2. Curiosamente, el complejo de evolución del oxígeno, y en particular el racimo de manganeso, es exclusivo de los fotótrofos oxigénicos. Nada parecido a eso se conoce en el resto de la biología. Jason Raymond y Bob Blankenship, sin embargo, tienen algunas ideas acerca de su procedencia. Han desarrollado nuevos métodos, que vimos antes por encima, para comparar las estructuras de las diferentes proteínas y han conjeturado que el racimo cuádruple de manganeso puede haber derivado de los racimos dobles de manganeso que se hallan en otras clases de proteínas. De hecho, cuando compararon el complejo de evolución del oxígeno con la proteína catalasa Mn, las similitudes en estructura resultaron razonablemente buenas.
La catalasa de manganeso es una proteína que convierte el peróxido de hidrógeno (agua oxigenada, vulgarmente), H2O2, en agua y oxígeno. Es una enzima detoxificante, puesto que el peróxido de hidrógeno es un compuesto dañino para los organismos. Según propusieron en primer lugar Bob Blankenship y Hyman Hartman, y recogieron después Raymond y el propio Blankenship, el racimo de manganeso de la catalasa Mn se incorporó en una bacteria fotótrofa anoxigénica algún tiempo después de que evolucionara la síntesis de clorofila. Este ensamblaje habría generado el primer organismo productor de oxígeno. Este organismo, sin embargo, habría dirigido una transferencia de dos electrones desde el peróxido para formar O2, en lugar de la más dificultosa transferencia de cuatro electrones desde dos moléculas de agua para formar también oxígeno, que es lo que vemos hoy en las plantas y las cianobacterias. En algún momento hubo una duplicación del racimo de dos Mn para formar un racimo de cuatro Mn, y esto permitió finalmente la evolución de la fotosíntesis oxigénica como la conocemos hoy.
No todo el mundo está satisfecho con esta hipótesis. La principal crítica es que el peróxido de hidrógeno era probablemente bastante raro antes de la evolución de la producción de oxígeno. John Allen y William Martin tienen otra idea y arguyen que el fotótrofo oxigénico primordial obetenía sus electrones de la fotooxidación de iones manganeso (en la forma química Mn2+) libremente disueltos en los primitivos océanos anóxicos. Los electrones obtenidos de esta manera habrían entrado en PSII, transfiriéndose luego a PSI y finalmente al CO2. Esto es similar a lo que ocurre en los modernos fotótrofos oxigénicos, con la diferencia de que la fuente de electrones estaba fuera de la célula. En este modelo, los iones Mn finalmente se desplazaron al interior de la célula y quedaron incorporados dentro de PSII, formando el complejo Mn. En el estadio final, el H2O devino la fuente última de electrones.
Redondearemos nuestra discusión sobre la evolución de la fotosíntesis oxigénica examinando la fase final del proceso, en que los electrones se combinan con el CO2 para fabricar materia orgánica. Este paso se consigue a través de un ciclo de reacciones bioquímicas conocido como el ciclo de Calvin-Bassham-Benson (o simplemente el ciclo de Calvin, para abreviar), donde la formación de materia orgánica a partir de CO2 es promovida por la enzima Rubisco, a la que ya conocimos antes en este capítulo. De hecho, Rubisco es la enzima más abundante del planeta. Aunque es característica de las cianobacterias productoras de oxígeno y de las plantas, también está presente en amplios grupos de bacterias fotosintéticas anoxigénicas (principalmente en las «bacterias púrpuras»), así como en toda clase de especies bacterianas que también convierten el CO2 en materia orgánica.
Uno de los aspectos fascinantes de Rubisco es que, por más que sea la enzima más abundante de la Tierra, no se puede decir que sea terriblemente buena haciendo su trabajo, al menos cuando la utilizan los organismos fotótrofos oxigénicos. Podemos identificar varias dificultades. Para empezar, Rubisco tiene una tasa de recambio extremadamente baja, procesando una molécula de sustrato cada 0,2 o 0,3 segundos, lo que la convierte en una de las enzimas más lentas conocidas; esto explica en parte su gran abundancia. En segundo lugar, su afinidad por el CO2 es bastante baja, aunque la biología ha aportado algunos remiendos para este problema, como veremos. Por último —y en mi opinión lo más curioso de todo—, Rubisco compite consigo misma de manera constante. Como ya hemos discutido, Rubisco fija CO2 en forma de compuestos orgánicos, en lo que se conoce como su actividad de carboxilasa (la c de Rubisco). También posee una actividad oxigenasa bastante increíble (la o de Rubisco) por la que el oxígeno reacciona con un compuesto intermediario para formar transitoriamente CO2 de nuevo, lo que esencialmente deshace la fijación de carbono previa. De modo que, como si no lograra decidirse, Rubisco se caracteriza por dirigir reacciones competidoras. Esto dista de ser un detalle trivial, porque se estima que la actividad oxigenasa de Rubisco reduce la tasa neta de fijación de carbono entre el 25% y el 40% en plantas como el arroz, el trigo y la soja.
El balance entre la actividad carboxilasa (la reacción favorecida) y la actividad oxigenasa (la reacción no favorecida) depende de la proporción de CO
2 a O
2 en el centro activo de Rubisco. Como cabría imaginar, cuanto mayor sea la proporción de CO
2 a O
2, más favorecida resulta la actividad carboxilasa. La sensibilidad de Rubisco al oxígeno también depende del tipo exacto de Rubisco que esté presente. Así, las Rubiscos utilizadas por los organismos anaeróbicos, como los fotótrofos anoxigénicos, tienden a poseer una alta actividad oxigenasa para la misma cantidad de CO
2, pero esto les da igual a estos organismos que raramente ven algo de oxígeno. Esta observación, sin embargo, abre una posible ventana para entender cómo los fotótrofos oxigénicos llegaron a usar Rubisco como su enzima fijadora de carbono. La historia es más o menos como sigue. Rubisco es una enzima fijadora de carbono ancestral que evolucionó mucho antes que los organismos productores de oxígeno. Esas antiguas Rubiscos tenían una actividad oxigenasa inherente;
[41] esto fue una propiedad accidental de la enzima, pero no tuvo consecuencias ya que no había oxígeno en el medio. La antigua Rubisco, surgida posiblemente en las bacterias púrpuras fotosintéticas anoxigénicas, fue adoptada por el primer fotótrofo oxigénico en un tiempo en que la concentración atmosférica de oxígeno era muy baja.
[42] A medida que la concentración de oxígeno fue aumentando, tanto en los entornos locales como en la atmósfera,
[43] los organismos desarrollaron diversas estrategias para enfrentarse a las limitaciones de la actividad oxigenasa de Rubisco. Estos remedios incluyen la evolución de Rubiscos menos sensibles al oxígeno y el desarrollo de una serie de estrategias para concentrar el CO
2 dentro de la célula, aumentando así la proporción de CO
2 a O
2. Las estrategias son ampliamente utilizadas hoy por las plantas y las cianobacterias.
Por tanto, ubisco aporta un bello ejemplo de cómo la evolución no conduce a una perfección de nivel superior. En este caso vemos que, una vez que Rubisco fue adoptada como la enzima fijadora de carbono por los fotótrofos oxigénicos, ese fue el camino que se tomó, y la evolución subsiguiente por selección natural ha actuado para minimizar la expresión de las imperfecciones de Rubisco, y no para encontrar un sustituto. Pero sus propiedades no están aún optimizadas para la tarea.
También vemos que tiene sentido ver la evolución de la fotosíntesis oxigénica en el contexto más amplio de la cambiante biosfera primitiva. Podemos ver, por ejemplo, de dónde se derivaron muchas de las partes que componen el proceso de la fotosíntesis oxigénica. En este contexto, no sorprende que la evolución del oxígeno llevara bastante tiempo, y se entiende por qué no era parte de la biosfera original, como examinamos en el último capítulo. La fotosíntesis oxigénica no fue, sin embargo, ensamblada como un rompecabezas a partir de varias piezas preexistentes. Muchas innovaciones biológicas exclusivas, como el racimo de manganeso y la biosíntesis de clorofila, por ejemplo, fueron parte integral de su evolución. ¿Podrían otras rutas haber conducido a la fotosíntesis oxigénica? Quizá. Esto, creo, es una cuestión fascinante, e imaginar rutas alternativas a la producción de oxígeno fotótrofa puede ayudarnos a visualizar cómo podría haberse desarrollado este proceso en otros lugares, más allá de la Tierra.
Capítulo 4
Cianobacterias: los Grandes Liberadores
Trate de imaginar algo tan profundo, tan fundamental, que haya cambiado el mundo entero. Piense en algo tan revolucionario que haya cambiado para siempre la química de la atmósfera, la química de los océanos y la naturaleza de la vida misma. ¿Qué tal la peste negra, el Renacimiento o la segunda guerra mundial? Fueron acontecimientos importantes en verdad, y todos cambiaron el curso de la cultura humana, pero su influencia fuera del dominio de nuestra especie fue pequeña. ¿Y qué hay de la extinción que mató a los dinosaurios hace 65 millones de años, o de la gran extinción pérmica de hace 250 millones de años, que barrió del mapa a quizá el 95% de todas las especies animales del planeta? Ahora nos estamos acercando. Cada una de estas grandes extinciones cambió realmente el curso de la evolución animal, pero aun así no alteraron de manera fundamental el tejido de la vida o la química de la superficie de la Tierra. ¿Qué fue capaz de alterarla, entonces?
La evolución de las cianobacterias lo fue. Estas modestas y minúsculas criaturas, a las que encontramos en el último capítulo, cambiaron todo por completo. Como ya hemos discutido, la evolución de las cianobacterias trajo por primera vez a la Tierra la producción biológica de oxígeno. Y esto acabó conduciendo a su vez a la acumulación de oxígeno en la atmósfera y a la evolución generalizada de los organismos que utilizan oxígeno. Analizaremos en detalle estos puntos en capítulos subsiguientes. La importancia de las cianobacterias, sin embargo, va más allá de eso. Como hemos visto, las cianobacterias fueron los primeros organismos fotosintéticos de la Tierra que utilizaron el agua como fuente de electrones. A diferencia del sulfuro, el Fe2+ y el H
2 usados por los organismos fotótrofos anoxigénicos, el agua está casi por todas partes en la superficie del planeta. Esto implica que la producción biológica en la Tierra ya no estaba limitada por la fuente de electrones (agua en este caso), sino más bien por los nutrientes y otros constituyentes menores que componen las células. En último término, el uso de agua en la fotosíntesis resultó en un incremento de las tasas de producción primaria en la Tierra, probablemente en un factor de entre diez a mil veces, como se apuntó en el capítulo 2.
[44] Por primera vez, la vida en la Tierra se volvió verdaderamente copiosa. Con la evolución de las cianobacterias, la Tierra se puso en marcha hacia su actual carácter de planeta verde.
[45]Las cianobacterias debieron de habitar todo tipo de lagos, estanques, corrientes y charcos en tierra firme donde el agua persistiera al menos durante periodos cortos, así como la entera capa superior de los océanos (llamada típicamente zona fótica). Si imaginamos a unos hipotéticos exploradores antiguos del sistema solar sondeando la Tierra primitiva, habrían necesitado un microscopio para encontrar evidencias de vida antes de la evolución de las cianobacterias.
[46] Después de ello, sin embargo, nuestros exploradores antiguos podrían haber encontrado vida abundante tomando imágenes de la Tierra desde la comodidad de su nave espacial, más o menos como hacemos nosotros con satélites en la actualidad. Allí donde era escasa antes de las cianobacterias, la materia orgánica se habría vuelto copiosa después de que evolucionaran. La degradación de esta materia orgánica conduce a los ecosistemas. Más materia orgánica conduce a ecosistemas más activos, y probablemente más complejos también. El incremento en la complejidad de los ecosistemas también habría resultado de la nueva disponibilidad de oxígeno y la subsiguiente evolución de los organismos que lo usaban. En conjunto, se habría dado un incremento en la diversidad biológica a medida que la abundancia tanto de materia orgánica como de oxígeno se iba haciendo disponible en entornos que abarcaban del mar a la tierra firme. Para resumir, la evolución de las cianobacterias fue un (o más probablemente
el) gran acontecimiento en la historia de la vida en la Tierra.
Mi introducción a las cianobacterias ocurrió justo después de que terminara mi tesis doctoral en la Universidad de Yale con Bob Berner. Bob poseía una paciencia infinita e hizo lo que pudo para convertir a un químico en algo parecido a un geocientífico. Aunque mi tesis se centraba en cómo la materia orgánica se reciclaba por los microbios en los actuales sedimentos marinos, el entrenamiento y estímulo de Bob consiguió que me quedara fascinado con los problemas de la historia de la Tierra, y en particular con la historia del oxígeno atmosférico. Ya mientras hacía la tesis me di cuenta de que, si quería comprender algo de esta historia, necesitaba aprender más sobre las cianobacterias productoras de oxígeno. Esto me condujo a una plaza de investigador posdoctoral con Dave des Marais en el centro de investigación Ames de la NASA en Palo Alto, California. Dave no sólo estaba interesado en la historia del oxígeno atmosférico, sino que también tenía en marcha un programa de investigación para estudiar las poblaciones modernas de cianobacterias.
Dave era también el mentor posdoctoral perfecto. Nos solíamos referir a él como «señor Mago», siempre desde el mayor respeto. Dave parecía saberlo todo y poder arreglar cualquier cosa. Nuestro trabajo de campo en las cianobacterias nos llevó a Baja Península, en México. El viaje de 1600 kilómetros a nuestra zona nos llevó, en camiones de la NASA sin marcas de identificación, a través de puestos fronterizos improvisados y cuestionables, a menudo atendidos por militares armados, hasta el hotel El Morro en Guerrero Negro. Una vez allí, convertíamos una o más de nuestras habitaciones en laboratorios (¡y dábamos propinas generosas al personal de limpieza cuando nos íbamos!) y montábamos nuestros tanques de incubación en el exterior y nuestro equipo eléctrico en el interior. Nuestras necesidades de suministro eléctrico estable estaban justo en el límite de lo que El Morro podía ofrecer. En casi todos los viajes, Dave se ocupaba de cambiar los fusibles en el cajetín principal del hotel (en el mejor de los casos) o, más probablemente, de reforzar algunas de las conexiones eléctricas más débiles. En una ocasión el hotel se quedó sin electricidad por completo. Dave halló la causa en una conexión floja de cable pelado que venía de la torre que suministraba electricidad al hotel. Con su calma característica para afrontar la catástrofe, Dave desenterró la conexión floja y la aseguró para que pudiéramos disfrutar de nuevo de un suministro fiable. En otra ocasión, el dispositivo de enfriamiento que yo había traído para controlar la temperatura de mis incubaciones se estropeó. Ningún problema. Dave reconectó el sistema de circulación de agua de forma que el sistema empezara a bombear agua cuando el tanque de incubación se calentara en exceso. Enganchando la bomba a un reservorio de hielo suministrado por la máquina de al lado,
[47] el problema quedó resuelto.
En cuanto empezamos el trabajo de campo, Dave se cambió su gorra de chapuzas por el sombrero de diplomático. Trabajábamos como invitados de la productora local de sal, la mayor de Norteamérica, y antes de cada campaña Dave explicaba cuidadosamente a los directivos de la compañía de qué modo nuestros resultados les ayudarían a optimizar su producción de sal. En aquellos días, nuestro permiso para investigar en México consistía en una carta firmada por el director de la empresa salinera donde se explicaba que nuestro trabajo era de extrema importancia para sus actividades.
Una vez dentro del recinto, emprendíamos el largo trayecto hasta nuestro lugar de trabajo. En un complejo masivo que cubre unos 500 kilómetros cuadrados, el agua del mar se transporta a través de una larga serie de estanques en los que se va evaporando bajo el calor y el viento del desierto. Al final la sal (NaCl) precipita en la concentrada salmuera. En las fases intermedias del proceso, cuando la salinidad del agua es de unas tres veces la del mar, se desarrollan vastas extensiones de unas espectaculares alfombras de cianobacterias (lámina 7). En la Tierra actual, las alfombras de cianobacterias se forman a menudo a altas salinidades, porque el agua muy salada elimina a la mayoría de los animales que de otro modo se comerían a las cianobacterias. En la Tierra primitiva, después de que evolucionaran las cianobacterias, pero mucho antes de que aparecieran los animales que se alimentan de ellas, las grandes extensiones de suelo marino en los litorales someros bañados por la luz solar debieron de constituir un hábitat adecuado. Estas regiones, junto al fondo de los lagos poco profundos, los ríos y los estanques, debieron parecerse mucho a los fondos de estos estanques actuales de Baja, México. En la Tierra primitiva, las cianobacterias formaban a menudo unas estructuras estratificadas conocidas como «estromatolitos», como siguen haciendo hoy en ciertos entornos (lámina 8).
[48] Nuestro trabajo era explorar las modernas alfombras de Baja, medir el nivel de actividad de las cianobacterias y aclarar la ecología del entorno en el que viven. Nuestro objetivo final era entender cómo las alfombras de cianobacterias podrían haber funcionado en la Tierra primitiva.
Exploramos muchos aspectos de esas alfombras, pero aquí me centraré en las propias cianobacterias. Estudiamos una especie conocida como
Microcoleus chthonoplastes, que predomina en las alfombras.
Microcoleus chthonoplastes es una cianobacteria de las llamadas filamentosas, en que las células se unen en fila india para formar largas cadenas llamadas «tricomas». Cualquier número de tricomas, desde dos hasta docenas de ellos, se apilan en haces que se fijan formando las alfombras. Los filamentos de cianobacterias se mueven arriba y abajo en los haces en respuesta a una variedad de estímulos como la luz, el oxígeno o los niveles de sulfuro. En las alfombras de Baja, estos haces se empaquetan en una red que recuerda de algún modo la consistencia del tofu. Dave des Marais hizo un bonito dibujo de una alfombra de Baja (figura 4.1) basada en imágenes de microscopio electrónico de transmisión. Si miramos esta imagen vemos que las cianobacterias en esta alfombra de Baja (y de forma similar a muchas otras alfombras microbianas de otros lugares) se concentran en una capa de menos de un milímetro de espesor. Se concentran ahí porque, hacia un milímetro de espesor en estas alfombras, la luz visible que usan se ha absorbido por completo.
[49] A medida que la luz disminuye, la abundancia de cianobacterias decrece, y en la base de la capa podemos ver una serie de filamentos mucho más pequeños orientados en su mayor parte horizontalmente en la alfombra. Son de color verde y contienen bacterioclorofila
a y
c. Representan un género de fotótrofos anoxigénicos conocido como
Chloroflexus o, más informalmente, como bacterias verdes no sulfurosas. Estos fotótrofos utilizan una luz con longitudes de onda que penetran por debajo de las cianobacterias, y usan esta luz para oxidar el sulfuro que producen los reductores de sulfato en las capas oscuras, profundas y libres de oxígeno de la alfombra. La ecología de esta alfombra es mucho más compleja de lo que indica mi simple descripción, pero este resumen presenta a los jugadores principales y muestra, básicamente, cómo puede estructurarse el ecosistema de una alfombra de cianobacterias.
Las dimensiones de esta alfombra están tan comprimidas que, para entenderla realmente, necesitamos instrumentos que puedan sondearla con una resolución de al menos 0,1 milímetros. Niels Peter Revsbech, de la Universidad de Aarhus en Dinamarca, abordó este desafío como parte de su proyecto de tesis doctoral, y desarrolló en los últimos años setenta los primeros microelectrodos para medir la distribución de oxígeno, sulfuro y grado de acidez (pH) en condiciones naturales. Estos electrodos minúsculos tenían en la punta un diámetro de meras micras (milésimas de milímetro, figura 4.2), y la necesaria resolución de escala fina. Niels Peter es un maestro en el diseño de electrodos, y ha seguido desde entonces desarrollándolos para uso en ecología. Estas innovaciones han espoleado importantes avances en la ecología microbiana al aportar una ventana para estudiar la química de las alfombras bacterianas (y otros ecosistemas naturales) a escalas casi comparables a las de los propios microbios. Nosotros sondeamos las alfombras de Baja con esos electrodos, y en la figura 4.3 se muestra un ejemplo de la distribución de oxígeno. A mi me resulta del todo asombroso que toda la acción con el oxígeno tenga lugar dentro de un espesor de menos de 2 milímetros. Separa el pulgar del índice 2 milímetros y trata de imaginar: en esa mínima distancia el oxígeno aumenta hasta unas cuatro veces la saturación del aire (¡eso es casi 1 bar de O2!) y cae hasta cero. Eso es durante el día. Cuando el Sol se pone, el pico en la concentración de oxígeno desaparece deprisa.
No mucho después de haber desarrollado los primeros microsensores de oxígeno, Niels Peter también dio con una forma hábil de determinar las tasas de producción de oxígeno en las alfombras microbianas.
[50] También aplicamos este método a nuestras alfombras, y en la figura 4.3 se muestra la distribución de las tasas de producción de oxígeno junto al perfil de su concentración. Esas tasas de producción de oxígeno son enormes, y están entre las más altas (por unidad de volumen) que se pueden encontrar en la naturaleza. Incluso si las integramos respecto a la profundidad para determinar las tasas de producción de oxígeno (que son equivalentes a las tasas de producción de materia orgánica por fotosíntesis oxigénica)
[51] por unidad de área (esto significa la tasa bajo un área dada de alfombra), encontramos que estas tasas también son altas. Por ejemplo, las tasas de producción de oxígeno integradas respecto a la profundidad, para los datos mostrados en la figura 4.3, ascienden a unos 20 milimoles de producción de oxígeno por metro cuadrado de superficie de alfombra y hora. Este valor es mucho mayor que las tasas que se encuentran en la mayor parte de los océanos globales, y es igualada o superada sólo en las áreas costeras de más alta productividad. ¡Y en las alfombras, toda esa producción de oxígeno ocurre en menos de un milímetro de profundidad! Esto es extraordinario. Las tasas de producción de oxígeno y las profundidades de su penetración varían de una alfombra a otra, pero como norma las alfombras de cianobacterias son extremadamente activas.
FIGURA 4.1 Ilustración de una típica alfombra de cianobacterias de Guerrero Negro, Baja California, México, basada en observaciones por microscopio electrónico de transmisión. La interfaz oxígeno-sulfuro se localiza durante el día a unos 0,8 mm de profundidad. Las letras se refieren a A, diatomeas; B, Spirulina (cianobacteria); C, Oscillatoria (cianobacteria); D, Microcoleus chthonoplastes (cianobacteria); E, bacterias no fotosintéticas; F, fragmentos de mucílago bacteriano; G, Chloroflexus (bacterias verdes no del azufre, capaces de fotosíntesis anoxigénica); H, Beggiatoa (bacterias no fotosintéticas oxidantes de sulfuro); I, intruso no identificado; J, cubiertas cianobacterianas abandonadas. Adaptado de Canfield y Des Marais (1991).
FIGURA 4.2 Diagrama de un microelectrodo a diferentes aumentos desarrollado por Niels Peter Revsbech, una herramienta estándar de la ecología microbiana. Redibujado a partir de Revsbech (1989).
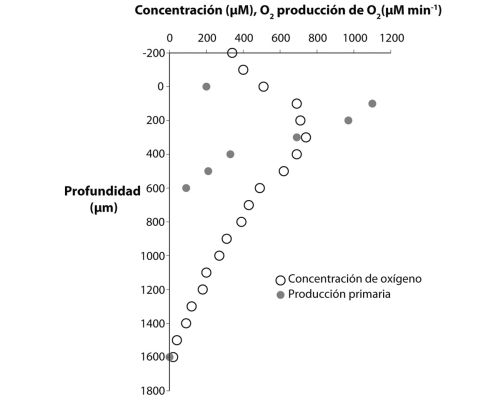
FIGURA 4.3 Distribución de oxígeno y tasa de producción de oxígeno en las alfombras microbianas de Baja, California, México. Datos de Canfield y Des Marais (1993).
Las cianobacterias no son raras. Están bien representadas en las alfombras microbianas como las que nosotros estudiamos en Baja y, si miramos con persistencia, podemos llegar a convencernos de que están casi por todas partes. Las encontramos en las playas de arena de Bornholm, como se describió en el capítulo 2. Pero eso es sólo el comienzo. También forman alfombras en el fondo de los lagos, estanques y arroyos, y son comunes en los suelos carentes de vegetación avanzada. Por tanto se hallan con particular abundancia, paradójicamente, en suelos áridos donde escasea el agua. Estas cianobacterias tienen la habilidad extraordinaria de sobrevivir a la sequía extrema en un estado letárgico, y de volver a la vida cuando el agua vuelve a estar disponible. Viaja al desierto de Mojave y coge un poco de arena. Es casi seguro que habrá una población apreciable de cianobacterias en tu mano. Viaja después a Uluru (antes llamado Ayers Rock) en Australia. Las arenas y suelos que rodean este gran monolito contendrán seguramente cianobacterias, pero mira más de cerca a la propia roca. Allí, y sobre todo justo debajo de la superficie de esa escultura natural de arenisca, viven abundantes poblaciones de cianobacterias. Viaja ahora a las regiones del Dry Valley de la Antártida, y encontrarás algo similar en los acantilados gigantes de Beacon Sandstone, frente al lago Vanda en el valle Wright de la Antártida. Afloramientos de granito y arenisca que contienen cianobacterias se hallan en otros entornos áridos y semiáridos que abarcan desde el norte del Transvaal en Sudáfrica hasta las tierras bajas del Orinoco en Venezuela.
Miremos a los océanos. Los científicos han estudiado las algas marinas durante siglos. Y, sin embargo, no hay una sola mención a las cianobacterias en el tratado clásico de Claude Zobell Microbiología marina, de 1946, como no eran las cianobacterias marinas descritas en el libro de texto de oceanografía biológica que yo usé en 1980. De hecho, por la época en que se escribió mi libro de oceanografía biológica (1977), las cianobacterias formadoras de haces del género Trichodesmium ya habían sido descritas (volveremos a estos interesantes organismos más abajo), y también se habían identificado otras cianobacterias pequeñas y cocoidales. Pero su novedad, aparentemente, era tanta que no habían entrado aún en las discusiones de los libros de texto. La situación cambió en 1979 cuando John Waterbury, de la Institución Oceanográfica Woods Hole, y sus colegas describieron la generalizada abundancia de las minúsculas cianobacterias del género Synechococcus. Resulta que, salvo en las zonas árticas, estas pequeñajas son comunes, con abundancias que pueden alcanzar el millón de células por litro de agua oceánica.
¿Cómo es posible que pasaran inadvertidas tanto tiempo? Durante décadas, el fitoplancton marino (que es el plancton fotosintético) se había recogido para su estudio mediante redes con un calibre de malla de 20 micras (0,02 milímetros). Pero las células de Synechococcus son muy pequeñas, con diámetros que oscilan de 0,8 a 1,5 micras. Por tanto, puedes pescar todo el fitoplancton que quieras, que las células de Synechococcus se escaparán de la red. John, sin embargo, recogió las células con filtros de un calibre de poro mucho menor (0,2 micras) y pudo así aislar estas minúsculas células de Synechococcus. Pero, como ocurre a menudo con los descubrimientos científicos, John no estaba buscando cianobacterias en absoluto, sino algo completamente distinto. Utilizaba una nueva técnica para cuantificar el número total de células bacterianas tiñéndolas con un pigmento fluorescente y contándolas bajo un microscopio mientras brillaban bajo la luz azul. Un buen científico hace controles (muestras sin tratar), y John vio de inmediato que algunas células brillaban con fluorescencia aunque no hubiera añadido el tinte. Algunos de los pigmentos fotosintéticos de los fotótrofos productores de oxígeno son fluorescentes por sí mismos y, puesto que aquellas células eran pequeñas, John sospechó que eran cianobacterias. Este resultó ser el caso, y los Synechococcus marinos habían sido descubiertos.
Pero esta no fue la última palabra acerca de las importantes poblaciones de cianobacterias en el mar. Más o menos al mismo tiempo que las células de Synechococcus eran identificadas como una especie abundante, se observaron otras poblaciones cianobacterianas de aspecto similar. Estos organismos presentaban una estructura interna algo distinta a la de Synechococcus, y sus pigmentos de clorofila eran también un poco diferentes; a falta de estudios más detallados, estos organismos fueron clasificados inicialmente como células de Synechococcus. Esto cambió en los últimos años ochenta cuando Penny Chisholm, del MIT, estaba usando una tecnología relativamente nueva llamada citometría de flujo para explorar la naturaleza de las poblaciones fotosintéticas del mar. Utilizando esta técnica, se pueden distinguir las poblaciones de acuerdo a su tamaño y a su capacidad de brillar con fluorescencia bajo luz de distintas longitudes de onda. Muchas poblaciones, como las de Synechococcus, generan señales distintivas, lo que constituye una excelente herramienta para cuantificar su tamaño de población. Aplicando la citometría de flujo, Penny pudo en efecto encontrar y cuantificar las poblaciones de Synechococcus. Pero también apareció otra población de células fluorescentes, y estas diferían de Synechococcus en muchos aspectos importantes. Primero, con diámetros de sólo 0,6 a 0,8 micras, estas celulitas eran mucho más pequeñas que las típicas células de Synechococcus. Aunque claramente eran cianobacterias, tenían pigmentos exclusivos que no se encuentran en Synechococcus, ni en ningún otro organismo fototrópico, dicho sea de paso. También eran abundantes, incluso más que Synechococcus, al menos en algunos lugares; y mientras Synechococcus solía preferir las regiones más superficiales y mejor iluminadas de la columna de agua, esta nueva población prefería zonas más profundas y oscuras. Sin duda eran las mismas células parecidas a Synechococcus que ya se habían descrito antes. Tras apreciar ciertas similitudes con un grupo de cianobacterias simbióticas del género Prochloron, Penny denominó a estas nuevas cianobacterias Prochlorococcus.
En unos 10 años, por lo tanto, nuestra comprensión de las comunidades fototrópicas marinas había cambiado por completo. Pero hay todavía más. Estas minúsculas cianobacterias, Synechococcus y Prochlorococcus, no sólo son abundantes, sino que pueden aportar hasta el 50% o más de la producción primaria en ciertas zonas del océano. Así que, por muy difíciles que sean de ver, las cianobacterias son muy importantes para el ciclo del carbono.
Y todavía hay más, porque desde luego no puedo dejar de explicar la ecología de Prochlorococcus en los océanos. Hago esto en parte porque es fascinante, y en parte porque, al pensar sobre el pasado lejano, podemos también imaginar que los organismos primitivos deben de haber vivido con adaptaciones a su entorno similares, e igualmente fascinantes. Para empezar, la especie Prochlorococcus tiene sólo unos 2000 genes, lo que la convierte en uno de los productores de oxígeno más escasos de genes que se conocen. Podría pensarse que cuantos más genes mejor, pues darían al organismo más flexibilidad y mejores opciones para adaptarse a las cambiantes condiciones del entorno. Prochlorococcus, sin embargo, utiliza una estrategia diferente. En lugar de cubrir todas las eventualidades con un gran genoma, estas cianobacterias se mantienen magras y ligeras, y modulan su genoma a las condiciones específicas del entorno.
La historia continúa reconociendo que Prochlorococcus no es sólo Prochlorococcus. Varias cepas diferentes de Prochlorococcus se han aislado de la naturaleza, y mientras que algunas están mejor adaptadas a condiciones de alta luminosidad, otras la prefieren baja, y otras más a las situaciones particulares de disponibilidad de nutrientes a diferentes profundidades del océano. Estos distintos ecotipos de Prochlorococcus, como los llama Penny Chisholm, se estratifican en la columna de agua marina, y esta estratificación también cambia con la latitud y la longitud respecto a su luz preferida y a su entorno químico. Un ejemplo de esa estratificación se muestra en la figura 4.4. Si empezamos por la distribución de Prochlorococcus cerca del ecuador, vemos que los ecotipos adaptados a la alta luminosidad prefieren la parte superior de la columna de agua, y los ecotipos de baja luminosidad prefieren aguas más profundas. Si nos movemos a latitudes más altas (48ºN), donde llega menos luz a la superficie del mar, los ecotipos de luz intermedia pueblan las capas superficiales y dominan en general. Aquí, el ecotipo adaptado a las mayores luminosidades está muy reducido en abundancia, y el ecotipo adaptado a la luminosidad más baja esta casi completamente ausente, pues aparentemente no hay bastante luz para que pueda competir con los demás ecotipos.
Podemos hacernos una idea de lo que hace un organismo, y de cómo se adapta al entorno, mediante una mirada a sus genes. Esto se debe a que los genes representan un plano de todos los distintos procesos que un organismo lleva a cabo. Hasta la fecha se han secuenciado los genomas de 12 tipos diferentes de Prochlorococcus. Cuando se comparan, se ve que hay 1270 genes compartidos por todos esos genomas. Estos genes representan el núcleo duro de lo que Prochlorococcus hace para ganarse la vida, e incluyen los genes que controlan la fotosíntesis y el metabolismo del carbono. Aparte de estos, hay cerca de otros 6000 genes variables que comparten algunos, pero no todos, los diferentes genomas. Estos dan a cada tipo de Prochlorococcus su estilo peculiar y determinan cómo cada uno se adapta específicamente a su entorno. La función de muchos de estos genes se desconoce, así que los detalles específicos de la adaptación al entorno son todavía nebulosos, pero el mensaje está claro. Mediante el trueque de ADN y la evolución de nuevos genes, Prochlorococcus ha construido genomas «de diseño» adaptados específicamente a las distintas condiciones ambientales que se pueden encontrar en los océanos. ¿Quién necesita un gran genoma si puede modular uno pequeño a las peculiaridades del entorno? No se sabe cuán general es esta estrategia entre los microbios de la naturaleza, pero el reconocimiento de la flexibilidad genómica de Prochlorococcus por Penny y sus colegas es, en mi opinión, una de las grandes revoluciones en la microbiología marina moderna.
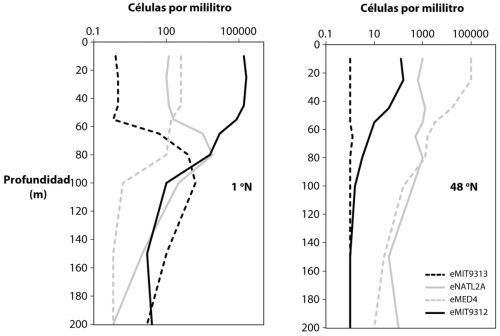
FIGURA 4.4 Distribución con la profundidad de diferentes cepas de Prochlorococcus a 1ºN y 48ºN de latitud en el océano Atlántico. Redibujado a partir de Jonson y colaboradores (2006).
Por si fabricar oxígeno no fuera suficiente, las cianobacterias también influyen en la química oceánica de otra manera esencial. Para entenderlo mejor, emprendo un crucero a bordo del Vidal Gómez, un viejo y desvencijado buque oceanográfico norteamericano comisionado a la Armada chilena. Viajamos al oeste desde Iquique, Chile, hacia un punto situado a unos 20 kilómetros de la costa. Los delfines juegan en la ola de proa, y los pelícanos buscan peces desde la distancia. Llegamos al sitio de estudio, y los delfines se dispersan; no hay diversión cuando el buque se queda parado. Me relajo y disfruto de las vistas, pero me sobresalta un banco de anchoas al romper la superficie al lado del barco. Mientras las ondas se asientan, miro hacia abajo y pienso que a sólo 100 metros bajo mis pies las aguas carecen por completo de oxígeno. Hemos venido a estudiar una de las mayores zonas de oxígeno mínimo (OMZ en sus siglas inglesas) que hay en los océanos del mundo. Estas zonas se encuentran al oeste de las costas de Perú y el norte de Chile, y más al norte, frente a las costas de Centroamérica y México, así como en el mar Arábigo del océano Índico. En estas áreas carentes de oxígeno, los microbios convierten el nitrato del agua marina en gas N2 y son responsables de retirar cerca de un tercio de todo el nitrato de los océanos. Sin reabastecimiento, el nitrato, un nutriente clave para la producción de las algas, se agotaría en los océanos en unos 5000 años.
Por fortuna, el reabastecimiento ocurre, y lo hace en muchos lugares de la superficie terrestre, pues la pérdida de nitrato en forma de N
2 no es sólo un fenómeno marino. El proceso de reabastecimiento se conoce como fijación de nitrógeno, por el que muchos tipos diferentes de microbios (más específicamente procariotas)
[52] convierten el gas N
2 en amonio para su uso por la célula. Se trata de un proceso muy intensivo en energía y complicado, dirigido por un complejo enzimático conocido como nitrogenasa. En tierra firme, estos procariotas viven a menudo en asociación simbiótica con las raíces de las plantas; las raíces de leguminosas como la soja son un buen ejemplo. En entornos acuáticos, las cianobacterias son los principales fijadores de nitrógeno. Esto tiene sentido, porque las cianobacterias obtienen mucha energía del Sol para llevar a cabo el proceso de fijación de nitrógeno. De forma paradójica, el oxígeno, el principal producto de las cianobacterias, es un veneno para la enzima nitrogenasa. La evolución ha generado varias soluciones muy astutas para este aparente dilema, y voy a esbozar algunas de ellas.
Varios tipos de cianobacterias filamentosas han desarrollado células especiales llamadas heterocistos. Están espaciados a intervalos semirregulares a lo largo del filamento, y la fijación de nitrógeno ocurre en esas células. Los heterocistos contienen múltiples capas de pared celular que restringen la difusión del oxígeno hacia dentro de la célula, y aunque poseen el fotosistema I, que produce la energía para conducir la fijación de nitrógeno, no tienen fotosistema II. Por tanto, y a diferencia de lo que ocurre en las células cianobacterianas normales, en los heterocistos no tiene lugar producción de oxígeno alguna. Al contrario, contienen proteínas especiales que consumen el oxígeno en solución, aportando así el entorno libre de oxígeno perfecto para la fijación de nitrógeno.
Las cianobacterias sin heterocistos tienen que encontrar otras formas de proteger a la nitrogenasa del oxígeno, y hay un montón de estrategias diferentes. Algunas cianobacterias que viven en alfombras microbianas, por ejemplo, sólo fijan nitrógeno por la noche, cuando el entorno de la alfombra se vuelve anóxico. Otras cianobacterias también restringen la fijación de nitrógeno a la noche, pero lo hacen en aguas ricas en oxígeno. En este caso, las cianobacterias respiran lo bastante rápido para retirar el oxígeno del interior de las células, de modo que la fijación de nitrógeno pueda proceder. La solución más sensacional, creo yo, es la de
Trichodesmium, que es seguramente el fijador de nitrógeno más importante de los océanos.
Trichodesmium es una cianobacteria filamentosa sin heterocistos, y a menudo forma unos haces impresionantes que se pueden observar a simple vista. De hecho, hay grandes agregados de
Trichodesmium fáciles de ver por satélite. De forma por completo contraria a la intuición,
Trichodesmium revela sus mayores tasas de fijación de nitrógeno a mediodía, cuando la intensidad de la luz alcanza el máximo. Esta sería normalmente la hora a la que las tasas de producción de oxígeno por fotosíntesis son máximas, puesto que la tasa de fotosíntesis suelen aumentar con la intensidad de la luz. No así con
Trichodesmium. Con alta luminosidad, estas cianobacterias desaceleran la fotosíntesis y usan la energía solar para conducir reacciones que utilizan oxígeno en la célula.
[53] Y al revés, en la penumbra de la mañana y el atardecer, la fotosíntesis se enciende y la fijación de nitrógeno se reduce al mínimo. Esta estrategia funciona cuando el suministro de luz solar es copioso, lo que explica que
Trichodesmium prefiera las aguas claras y las latitudes tropicales (¿y quién no?).
Hasta ahora hemos explorado varios aspectos de la ecología y la fisiología de las cianobacterias, pero hemos ignorado a propósito las plantas y las algas. ¿Qué hay de ellas, puesto que también hacen oxígeno? De hecho, ¿podría haber alguna relación entre las diminutas cianobacterias a las que tenemos que esforzarnos por ver y las plantas y algas que casi definen nuestro mundo moderno? Pues bien, resulta que esa relación es muy estrecha, y revela un maravilloso accidente en la historia de la vida. Hace mucho, mucho tiempo, una cianobacteria fijó su residencia dentro de una célula eucariota. Esto fue una relación beneficiosa para ambas partes (conocida normalmente como simbiótica), en la que la célula eucariota probablemente logró alimento de la cianobacteria, y la cianobacteria probablemente obtuvo refugio en el interior del eucariota. Pasando el tiempo evolutivo, el eucariota tomó el control de la cianobacteria, que fue perdiendo gran parte de su propia maquinaria metabólica. La cianobacteria perdió lentamente su identidad como un organismo independiente y se convirtió en el cloroplasto del alga eucariótica ancestral. Esta idea fascinante fue concebida originalmente por el botánico ruso Konstantin Sergeevich Merezhkovsky en 1905. La idea se perdió hasta ser redescubierta y popularizada por Lynn Margulis muchas décadas después. Desde entonces se ha demostrado correcta con las técnicas modernas de la biología molecular, que han mostrado clara y netamente que el cloroplasto contiene ADN cianobacteriano.
Debemos mucho a nuestras pequeñas amigas productoras de oxígeno. Nos han dado oxígeno para respirar, y se han asegurado de que haya abundante nitrato en el océano para mantener las grandes cadenas alimentarias de los mares. Se han comprometido en asociaciones con los primitivos eucariotas para darnos las algas y, finalmente, las plantas, y hoy se encuentran poblando innumerables entornos distintos con fascinantes adaptaciones a cada uno de ellos. Para mí es difícil pensar acerca de la vida en la Tierra sin guiñar un ojo a las cianobacterias y agradecerles que hayan hecho posible tan gran parte de ella.
Capítulo 5
¿Qué controla el nivel de oxígeno atmosférico?
Aspiración, expiración, aspiración, expiración. Bonito y relajado. Cada uno de nosotros hace esto quizá 20 000 veces al día y raramente le dedicamos un solo pensamiento. Yo, en cambio, pienso más sobre la respiración si viajo a Santa Fe, Nuevo México, a una altitud de 2100 metros sobre el nivel del mar. Justo después de llegar subo jadeando un piso de escaleras y, durante un corto paseo por las colinas, resoplo y resuello mucho más de lo habitual. A esta altitud, la presión atmosférica sólo alcanza el 77% de la típica del nivel del mar, lo que significa que, para la misma aspiración, sólo tomamos el 77% del oxígeno que allí. En el techo del mundo, la cima del monte Everest en los Himalayas, la altitud es de unos impactantes 8848 metros, y aquí una pulmonada de aire contiene sólo el 31% del oxígeno que tendría al nivel del mar. Sólo los mejor entrenados y mejor adaptados pueden sobrevivir con tan poco oxígeno, y aún así solo por un tiempo corto. La mayoría de los escaladores hacen la última parte del ascenso con una bombona de oxígeno. Algunos no la llevan, y muchos de estos mueren en el intento. La cima del Everest claramente pone a prueba los límites de la resistencia humana.
Como se ve, la cantidad de oxígeno en el aire es importante. El contenido de oxígeno en el aire es actualmente del 21%, y haríamos bien en preguntarnos por qué la concentración es esa y no otra.
[54] También podríamos preguntar si esa concentración ha cambiado con el tiempo. Examinaremos la historia del oxígeno atmosférico en capítulos posteriores, pero aquí nos ocuparemos con la cuestión más fundamental de por qué hay oxígeno en la atmósfera en absoluto. A través de esa discusión intentaremos identificar los principales procesos que controlan la concentración de oxígeno.
Pues vaya novedad, pensará el lector. Cualquier colegial sabe que el oxígeno proviene de la fotosíntesis. Por eso tenemos oxígeno en el aire, así que ¿cuál es el problema? Cierto, pero haría falta un colegial realmente listo para concebir el siguiente experimento. Pon una planta en un recipiente cerrado y mira cuánto oxígeno se acumula durante el día.
[55] Apunta la cantidad. Ahora, observa qué pasa por la noche. Probablemente verás que, durante la noche, la planta utiliza casi tanto oxígeno al respirar como el que produjo durante el día.
[56] Estamos casi donde empezamos: pero no exactamente, y ese es el punto.
Repasemos las medidas que hemos hecho. El nivel de oxígeno es probablemente un poco más alto, y si prolongamos el experimento por unas semanas veremos seguramente que se va acumulando algo de oxígeno. Esto se debe a que la planta ha crecido. Recuerda que el oxígeno es un subproducto de la fotosíntesis y que la producción de oxígeno viene compensada por la producción del material de la planta. Cuanto más material de la planta se genere, más oxígeno se produce. En nuestro experimento, si todo el material de la planta se utiliza para respirar, no se acumulará nada de oxígeno, pero si la planta crece, este crecimiento representa material de la planta que no ha respirado. En otras palabras, si el oxígeno no se usa para que el material de la planta respire, se acumula en el recipiente. De esta forma, el crecimiento de la planta puede igualarse a la acumulación de oxígeno. Así de simple; ¿o no?
Intentemos el siguiente cálculo. Actualmente hay en la atmósfera 3,7×1019 moles de oxígeno. Es un número grande. Si lo enfriáramos hasta el estado líquido, formaríamos una capa de oxígeno líquido de 6 centímetros que cubriría la superficie entera del planeta. Con una combinación de imagen por satélite y mediciones desde tierra, se estima que la tasa neta de producción primaria en el planeta, que es más menos lo mismo que el crecimiento combinado de las plantas, algas y cianobacterias, es de unos 8,8×1015 moles de carbono al año. Si comparamos esta tasa de producción primaria neta con la masa de oxígeno en la atmósfera, podemos calcular que todo el oxígeno de la atmósfera podría generarse en tan sólo 4200 años. Este cálculo implicaría que el contexto de oxígeno de la atmósfera debería ser inestable en intervalos de tiempo bastante cortos, y que un ligero desequilibrio entre la producción de oxígeno y la respiración nos podría provocar unas concentraciones de oxígeno tan bajas como las del monte Everest, o alternativamente unos niveles muy altos.
Pero no te preocupes, que eso es sólo la mitad de la historia. Volvamos a nuestro experimento del recipiente. El oxígeno se acumula en el envase mientras la planta esté viva y creciendo. Pero ¿qué pasa si la planta se muere? Al igual que ocurre en la pila de compost del patio trasero, todo tipo de bacterias y hongos descompondrán el material de la planta muerta, usando oxígeno para hacerlo. En conjunto, ya sea en el mar o en tierra firme, se estima que el 99,9% de la producción primaria de la Tierra se descompone. Los restantes trocitos quedan enterrados como materia orgánica no reactiva en sedimentos marinos y de agua dulce, que pueden después solidificar como rocas. De hecho, esta materia orgánica enterrada en sedimentos y luego transformada en roca es la única que se escapa de reaccionar con el oxígeno. El enterramiento de esta materia orgánica supone por tanto una fuente neta de oxígeno para la atmósfera. De modo que las plantas y las cianobacterias producen el oxígeno, pero este se acumula sólo porque parte de la materia orgánica original producida por fotosíntesis es enterrada y preservada en sedimentos.
[57] Un trozo de carbón representa una fuente de oxígeno para la atmósfera, como también un barril de crudo, los fósiles orgánicos y toda la materia orgánica finamente dispersa que confiere su bello color negro a las pizarras primitivas que tanto me gusta investigar.
[58]Hay otra fuente de oxígeno que tenemos que considerar. Como vimos en el capítulo 2, un proceso microbiano anaeróbico llamado «reducción del sulfato» respira materia orgánica convirtiendo el sulfato en sulfuro. Este proceso es bastante común en la naturaleza. Podemos encontrar bacterias reductoras de sulfato en los huevos podridos, en nuestros intestinos y hasta en nuestros dientes. Pero son más prominentes en cuencas relativamente aisladas como el mar Negro y la cuenca de Cariaco frente a la costa de Venezuela, en muchos de los fiordos escandinavos en que la circulación del agua está restringida y en la mayoría de los sedimentos marinos a las profundidades en que el oxígeno se ha consumido por respiración. Estas profundidades de los sedimentos oscilan entre sólo un milímetro o dos, en entornos cercanos a los márgenes continentales, hasta varios centímetros o más, cuando recogemos fango mar adentro y donde los contenidos orgánicos son relativamente escasos. Si hay hierro por la zona —y lo suele haber—, el sulfuro reacciona con él formando un mineral llamado pirita (fórmula química FeS
2), que ya encontramos en el capítulo 2. En los sedimentos modernos, este «oro del tonto» (
fool’s gold) se halla típicamente en forma de unos hermosos racimos como frambuesas microscópicas (llamados framboides, del francés
framboise) con un diámetro entre 5 y 50 micras (figura 5.1). En las rocas antiguas, la pirita suele encontrarse en forma de cubos de un dorado reluciente, pero en cualquier caso el sulfuro (y el hierro) atrapado como pirita es muy sensible al oxígeno, y vuelve a convertirse en sulfato si se expone a él. Como el sulfuro se había formado a partir del poder reductor de la materia orgánica, si no reacciona y queda enterrado en sedimentos, la deposición de pirita también representa una fuente de oxígeno para la atmósfera.
[59] Como veremos en capítulos posteriores, el enterramiento del carbono orgánico ha sido la principal fuente de oxígeno atmosférico durante los últimos cientos de millones de años, pero durante algunos intervalos más antiguos la principal fuente de oxígeno fue el enterramiento de pirita.
Resulta extraordinario que estos controles de la concentración de oxígeno atmosférico fueran elucidados ya en 1845 por el químico e ingeniero de minas francés Jacques Joseph Ebelmen; comprendió estos procesos apenas 70 años después de que se descubriera el oxígeno como elemento y de que su producción se vinculara a la fotosíntesis, como se discutió en el capítulo 3. De manera trágica, Ebelmen murió a la temprana edad de 37, pero durante su corta y distinguida carrera pensó con profundidad acerca de qué procesos influyen en el reciclado del oxígeno en la atmósfera. Escribió reacciones químicas que describían los controles del oxígeno atmosférico, en un tiempo en que esas representaciones eran bastante nuevas. Ebelmen llegó a la conclusión, como apunté antes, de que el enterramiento tanto la pirita como del carbono representan fuentes de oxígeno para la atmósfera. De esta forma pudo distinguir entre el reciclado a corto plazo del oxígeno, definido por la fotosíntesis y la respiración de los organismos, y su reciclado geológico a largo plazo, regulado por el ciclo de las rocas. El ciclo de las rocas, como exploramos en el capítulo 1, incluye el enterramiento de los sedimentos, la transformación de los sedimentos enterrados en rocas y la posterior elevación tectónica de esas rocas, que entonces quedan sometidas a la erosión física y química. Los productos de esas erosiones son transportados por los ríos a los océanos, donde vuelven a formar sedimentos.
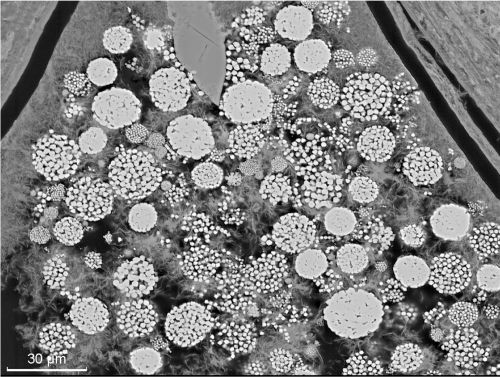
FIGURA 5.1 Numerosos framboides de pirita en una fina sección petrográfica de una pizarra. Foto cortesía de Eric Condliffe, de la Universidad de Leeds.
Estos conceptos de control de oxígeno permanecieron en estado mayormente latente durante 130 años, hasta ser redescubiertos en la década de 1970 de forma independiente por Bob Garrels, Ed Perry y Dick Holland. Tendré mucho más que decir sobre estos científicos y sus contribuciones en capítulos posteriores.
Hasta ahora tenemos bajo control las fuentes de oxígeno, pero ¿qué pasa con todo este oxígeno una vez liberado a la atmósfera? Si puedes, date una vuelta con el coche en un bonito día de verano por un afloramiento de rocas sedimentarias, pizarras a ser posible. Si vives cerca de una zona montañosa, puedes encontrar el afloramiento perfecto en un tajo de carretera o junto a un río, o simplemente en la ladera de una colina. Si vives en una zona plana como yo, tal vez tengas que esperar hasta salir de vacaciones. En cualquier caso, echa una mirada detenida a las pizarras; usa una lupa si la tienes. Fíjate en las capas más externas. Seguramente serán desmenuzables, y pueden aparecer decoloradas. Es probable que veas manchas de óxido de hierro y, si prestas atención, este óxido puede reemplazar a lo que parecen cubos o lentes de pirita en la roca. Si tienes un martillo de geólogo, aparta el material desmenuzable y trata de encontrar algo de roca fresca. La roca fresca será probablemente más oscura, y si penetras lo bastante verás brillar la pirita fresca. Sin más que comparar la roca erosionada con la fresca, habrás descubierto lo que retira gran parte del oxígeno de la atmósfera. El oxígeno reacciona con la materia orgánica y la pirita en las rocas antiguas, una vez que estas han vuelto a aflorar a la superficie de la Tierra. Esta materia orgánica y pirita, de hecho, fue una fuente de oxígeno al ser enterrada y convertirse en roca. Su oxidación final representa un sumidero de oxígeno y completa el ciclo.
Hay otra ruta de retirada de oxígeno que debemos discutir. En el capítulo 2 discutimos cómo los gases reducidos químicamente como el H2, el H2S y el SO2 se liberaban al entorno de superficie desde los volcanes, y cómo probablemente aportaban energía a los ecosistemas primitivos antes de que evolucionara la producción de oxígeno. Después de que surgiera la producción de oxígeno por las cianobacterias, estos gases volcánicos devinieron sumideros de oxígeno. El sulfuro y el SO2 reaccionan con el oxígeno para formar sulfato, mientras que el H2 reacciona con el oxígeno para formar agua. La figura 5.2 presenta un dibujo esquemático que resume estos puntos y muestra las fuentes y sumideros de oxígeno en el ciclo geológico del O2.
FIGURA 5.2 Los principales procesos que controlan las concentraciones atmosféricas de oxígeno. El oxígeno reacciona con los gases volcánicos, y también con el carbono orgánico y los compuestos de azufre durante la erosión, mientras que es liberado a la atmósfera por el enterramiento del carbono orgánico y la pirita en los sedimentos. Modificado de Canfield (2005).
Saber todo esto es esencial para entender cómo se recicla el oxígeno, pero sigue sin explicar cómo se controlan sus concentraciones. En concreto, ¿podemos identificar procesos en el ciclo del O2 que verdaderamente regulen los niveles de oxígeno? La respuesta es sí. El truco es reparar en que las tasas de muchos de los procesos que producen y consumen oxígeno tienden a conducir a unos niveles de oxígeno estables. Esto puede parecer bastante oscuro, pero no te preocupes. Ilustraré el principio antes de entrar en más detalles sobre los mecanismos específicos.
Imagina una bañera muy profunda con un grifo y un desagüe (figura 5.3). Cuanta más agua haya en la bañera, más deprisa saldrá por el desagüe. Abre el grifo y deja que entre agua en la bañera. El agua alcanzará un nivel tal que saldrá por el desagüe a la misma velocidad por la que entra del grifo. Ahora echa un gran cubo de agua al baño. El nivel subirá de repente y hará que el agua se vaya por el desagüe más rápido. Tras esa elevación inicial, el nivel caerá hasta el mismo punto de antes, el punto en el que la entrada iguala a la salida. Esto se conoce como retroalimentación negativa (o feedback negativo), y estabiliza el sistema. Ahora saca de la bañera un gran cubo de agua, y pasará lo contrario. La velocidad de salida por el desagüe se ralentiza hasta que se alcanza de nuevo el nivel original. El sistema ha alcanzado un estado estable, y ese estado se mantiene gracias a la simple retroalimentación.
FIGURA 5.3 La metáfora de la bañera sobre las varias formas en que se regula la concentración de oxígeno atmosférico; en este caso, los niveles de oxígeno se representan como la altura (h) del agua en la bañera. El texto da una descripción del funcionamiento.
Si abrimos más el grifo, se alcanzará un nuevo nivel de agua más alto. El nivel de agua sube y más agua sale por el desagüe hasta compensar la mayor entrada. Esto es otro ejemplo de retroalimentación negativa estabilizadora. También se alcanzará un nivel de agua más alto si restringimos el desagüe. O al revés: si aumentamos el desagüe o reducimos la apertura del grifo, se alcanzará un nivel de agua más bajo. Ahora metamos un tubo en la bañera y absorbamos agua a velocidad constante (un sifón). Veremos el nivel del agua caer a un nuevo nivel en que la tasa total de salida por el desagüe y el sifón vuelve a igualar a la entrada por el grifo. Por último, añadamos un flotador conectado al grifo. El flotador funciona más o menos como el de la cisterna del váter (aunque no es tan sensible) y, cuanto más alto sea el nivel del agua, hará que más lenta salga por el grifo. El flotador ayuda a definir un nivel de agua máximo en la bañera. Con este mecanismo del flotador, la entrada de agua se reduce cuando el nivel sube. Se trata de un sistema más de retroalimentación negativa que actúa para estabilizar el nivel de agua en la bañera.
Este simple sistema, por supuesto, es una metáfora de cómo se controla el oxígeno atmosférico y, como veremos a continuación y en capítulos posteriores, contiene todos los principios que necesitamos para comprender la regulación del oxígeno. Como ocurre con la concentración de oxígeno atmosférico, la bañera alcanza un equilibrio dinámico en el que las entradas compensan a las salidas. El equilibrio dinámico es posible gracias a las retroalimentaciones negativas. El sistema también contiene un mecanismo de pérdida de agua, el sifón, que opera con independencia de cualquier retroalimentación. Esta ruta de retirada de agua influye en el nivel de la bañera, pero no desestabiliza el sistema necesariamente. En otras palabras, un equilibrio dinámico sigue siendo posible con el sifón funcionando, aunque con un nivel más bajo del que se alcanza con el sifón apagado.
Las retroalimentaciones positivas también son posibles. Son retroalimentaciones desestabilizantes. En nuestra metáfora del baño, un ejemplo de retroalimentación positiva podría ser que el flotador estuviera amañado de manera que el flujo de entrada aumentara cuando el nivel de agua sube. Con esta retroalimentación positiva, la bañera acabaría desbordándose. Como veremos, en la regulación del oxígeno también hay posibles retroalimentaciones positivas, pero no son los controles predominantes de la concentración de oxígeno. El nivel de oxígeno se estabiliza por retroalimentaciones negativas. Veamos algunas.
Empecemos por las rutas de retirada del oxígeno, y en concreto por la más simple de todas: la liberación desde el manto de gases reactivos con el oxígeno. Me refiero en particular a gases como H2, CO (monóxido de carbono), SO2 y H2S, que son los análogos directos del sifón en nuestro ejemplo de la bañera. Estos gases son sumideros de O2 y se añaden a la atmósfera de manera independiente de cualquier retroalimentación que opere en el ciclo del oxígeno. Su velocidad de entrada en la atmósfera depende de la tectónica y de las agitaciones internas de la Tierra. Como veremos en el capítulo 7, estos gases han tenido un papel esencial en la regulación del oxígeno atmosférico durante algunos periodos de la historia de la Tierra.
¿Qué hay de otras rutas de retirada del oxígeno, como la oxidación del carbono orgánico y la pirita en las rocas sedimentarias, de las que ya hablamos antes? Estas son equivalentes al desagüe de nuestra bañera. Los experimentos han mostrado que la velocidad de oxidación tanto de la pirita como del carbono orgánico dependen en realidad de la concentración de oxígeno en la atmósfera, con una oxidación más rápida a mayores niveles de oxígeno. Esto constituye un mecanismo potencial de retroalimentación negativa en la disminución de oxígeno. Si el oxígeno se vuelve demasiado bajo, la velocidad de su retirada por oxidación de carbono orgánico y pirita disminuye. Como estudiante de postgrado de primer año en Yale, presencié un debate sobre este punto entre Karl Turekian, un profesor de Yale, y Bob Garrels,
[60] a quien ya hemos encontrado brevemente en este capítulo. Karl Turekian era un gladiador, un científico inspirador que se crecía en la confrontación. Bob Garrels era un sabio de la geoquímica, de habla suave y amables formas. Garrels era quien daba la charla, y puntualizó que la concentración de oxígeno actuaba probablemente como una retroalimentación negativa sobre las tasas de erosión química del carbono orgánico y la pirita. Entonces Karl saltó de la silla y, saboreando una buena pelea, bramó que eso no podía ser cierto. La velocidad de erosión química está controlada por la tasa de elevación geológica y por la tasa a la que la nueva roca queda expuesta en la zona de erosión en la superficie de la Tierra. Una vez allí, prosiguió Karl, todo el carbono orgánico y la pirita será oxidado antes de que la roca erosionada sea transportada a los ríos y de vuelta al mar. Yo, viendo subir la temperatura de la discusión, estuve tentado de esconderme debajo del escritorio. Garrels, sin embargo, era la viva imagen de la serenidad budista; sonrió y dijo: «Bueno, Karl, esa es tu opinión», y continuó con su charla.
Lo cierto es que Karl, como de costumbre, había suscitado un buen argumento. Si la materia orgánica y la pirita se oxidan por completo durante la erosión y el transporte río debajo de camino hacia el mar, no habrá retroalimentación del oxígeno sobre la velocidad de oxidación de la materia orgánica y la pirita. De hecho, la trascendencia de esta retroalimentación podría muy bien depender del contenido real de oxígeno en la atmósfera. Volvamos al afloramiento de rocas que discutimos antes, aquel donde observamos la roca erosionada en la superficie, y donde la roca prístina emergió con sólo escarbar un poco. En vez de limitarnos a observar la roca, recojamos muestras a intervalos regulares de su superficie erosionada y también de la roca fresca que yace bajo ella. Nos llevamos las muestras de vuelta al laboratorio y medimos su contenido en pirita y materia orgánica. Steven Petsch, ahora en la Universidad de Massachusetts, hizo eso como parte de su tesis doctoral con Bob Berner en Yale, unos 10 años después de que yo acabara la mía también con Bob. Algunos de los resultados de Steve se muestran en la figura 5.4. Hay dos conclusiones principales que emergen del trabajo de Steve. Una es que la pirita se oxida mucho más rápido que la materia orgánica. La otra es que, aunque la materia orgánica se oxida más lentamente que la pirita, en muchos casos (pero no en todos) aparece completamente oxidada en las capas superficiales del afloramiento.
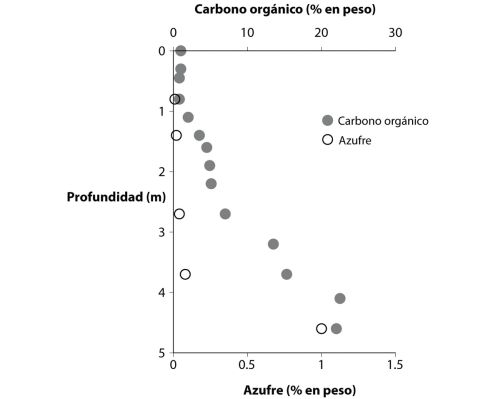
FIGURA 5.4 Concentraciones de carbono orgánico y azufre pirítico a través de un suelo moderno en formación en las pizarras de Woodford de las montañas Arbuckle, condado de Murray, Oklahoma, Estados Unidos. Redibujado de Petsch y colaboradores (2000).
Estas observaciones formaron la base de un elegante modelo de oxidación de materia orgánica/pirita formulado por Ed Bolton, experto modelador y uno de los colegas de Bob Berner en Yale. El modelo de Ed es bastante complejo e incluye una variedad de parámetros como la concentración de oxígeno atmosférico, la velocidad de erosión, la porosidad de la pizarra (lo que significa básicamente el volumen de espacio vacío, o agujeros en la pizarra), la dinámica del flujo de agua y la cinética de oxidación de la pirita y el carbono orgánico. Los resultados, sin embargo, están claros. Dada la gama normal de velocidades de erosión y porosidad del suelo, la materia orgánica se oxidará totalmente durante la erosión, siempre que el nivel de oxígeno atmosférico sea el 25% del actual o mayor. Con niveles inferiores de oxígeno, sin embargo, o con tasas de erosión mucho más altas, como las típicas de los terrenos montañosos, por ejemplo, la concentración de oxígeno sí debería afectar al grado de oxidación de la materia orgánica. Por tanto, el oxígeno atmosférico podría haber aportado una retroalimentación negativa sobre la oxidación de la materia orgánica en el pasado, cuando los niveles de oxígeno eran considerablemente menores que hoy, o cuando las tasas de erosión eran considerablemente más altas. Una retroalimentación negativa de la concentración de oxígeno sobre la oxidación de la pirita es también posible, pero sólo a concentraciones de oxígeno extremadamente bajas; exploraremos esto en el capítulo 9.
La discusión se ha centrado hasta ahora en la eficiencia de la oxidación de la materia orgánica y la pirita, pero la cantidad de materia orgánica disponible para la oxidación debe tener también un papel. De un modo simple, cuanta más materia orgánica y pirita esté disponible para oxidarse, mayor será el sumidero de oxígeno. La cantidad disponible dependerá de varios factores. Uno es la tasa de levantamiento de los continentes, puesto que controla la velocidad a la que las rocas se exponen a la erosión. Otro factor es la concentración de carbono orgánico y pirita en las rocas sometidas a erosión, y esto dependerá en gran medida del tipo de rocas que han quedado expuestas. Bob Berner ha pensado profundamente en esto, y ha concebido una forma en que la concentración de oxígeno atmosférico puede regularse según este principio. Bob propone que hay un acoplamiento estrecho entre el enterramiento de carbono orgánico y pirita en los sedimentos, que representa una fuente de oxígeno, y la subsiguiente disponibilidad de esos mismos sedimentos para la erosión y la oxidación, que representa un sumidero de oxígeno.
El razonamiento es el siguiente: imagina que, por alguna razón, hay un periodo en que se forman sedimentos muy ricos en carbono orgánico y pirita. El enterramiento de estos sedimentos representaría una fuerte fuente de oxígeno para la atmósfera. Sin embargo, estos sedimentos, o al menos muchos de ellos, serían muy propensos a la oxidación física y química en unas escalas de tiempo relativamente cortas.
[61] Esto se debe a que esos sedimentos (y las rocas que forman) están sujetos a la exposición durante los cambios del nivel del mar. También están sujetos al levantamiento y la erosión como resultado de varios procesos tectónicos como la subducción del suelo marino y las colisiones entre continentes.
[62] Por tanto, el
reciclado rápido de estas rocas ricas en carbono orgánico y pirita hacia la zona de erosión y oxidación generaría pronto un fuerte sumidero de oxígeno. El resultado final es que la fuerte fuente de oxígeno representada por el enterramiento inicial de estas rocas se compensa por el fuerte sumidero de oxígeno que supone su posterior oxidación. De forma inversa, si hay un periodo de deposición de sedimentos con bajo contenido en carbono orgánico y pirita, la influencia de esta débil fuente de oxígeno se amortigua por el débil sumidero de oxígeno generado por el reciclado rápido y subsiguiente oxidación de este mismo sedimento. Esto evita que el oxígeno baje demasiado. El resultado final del reciclado rápido de sedimentos es evitar que las concentraciones de oxígeno atmosférico se eleven o reduzcan demasiado. Hay que señalar que este mecanismo para estabilizar la concentración de oxígeno no representa una retroalimentación sobre la concentración de oxígeno; no depende del contenido de oxígeno en la atmósfera. En vez de eso, es un mecanismo de estabilización basado en el acoplamiento entre el enterramiento de sedimentos, la tectónica y la erosión química.
Las retroalimentaciones y controles descritos arriba se refieren a los sumideros de oxígeno. También podemos identificar varias retroalimentaciones asociadas a la producción de oxígeno. Un grupo importante de retroalimentaciones tiene relación con la influencia de la anoxia oceánica sobre la regulación del oxígeno.
[63] Empezaremos esta discusión examinando el sedimento de la zona profunda del mar Negro. El mar Negro es la mayor cuenca anóxica del mundo; por debajo de 70 u 80 metros de profundidad, el agua está libre de oxígeno, y si recoges agua por debajo de 120 metros, apesta a sulfuro, el producto de la reducción del sulfato. Este tipo de cuencas sulfúricas se conocen como cuencas euxínicas, precisamente por el nombre griego del mar Negro,
Pontus Euxinus.
Los sedimentos que se depositan en las aguas euxínicas del mar Negro están desprovistos de animales. Además, están finamente laminados y llenos de carbono orgánico y sulfuro de las piritas, mucho más que del que encontramos en los fangos ordinarios que rodean las costas de Dinamarca, o en cualquier lugar cercano a las playas que la gente frecuenta en vacaciones. De hecho, las tasas de enterramiento de carbono orgánico y pirita aparecen acentuadas cuando la columna de agua se vuelve euxínica.
Antes de continuar, sin embargo, hay un detalle importante que necesitamos considerar. No todos los tipos de anoxia en la columna de agua conducen a la clase de condiciones euxínicas que encontramos en el mar Negro. Por ejemplo, muchos lagos en la zona del medio-oeste de Estados Unidos donde yo me crié (y en otros lugares también) se vuelven estacionalmente anóxicas en sus aguas profundas. Sin embargo, estos lagos suelen mostrar bajas concentraciones de sulfato, de modo que sólo pueden sustentar tasas limitadas de reducción de sulfato. Por tanto, cuando estos lagos se vuelven anóxicos, en vez del sulfuro se acumula el hierro disuelto (llamado hierro ferroso, veremos mucho más de esto en el capítulo 7). Esta situación fue también muy común en los océanos durante ciertos periodos del pasado geológico, como se explora en capítulos subsiguientes. Si recuerdas las aguas carentes de oxígeno junto a la costa de Chile que discutimos en el último capítulo, ni el hierro ni el sulfuro se acumulan allí, sino que en esas aguas domina la química del oxígeno. Lo importante, sin embargo, es que todos los tipos de anoxia de la columna de agua conducen a tasas reducidas de descomposición de la materia orgánica (comparadas con las descomposiciones que usan oxígeno), lo que conduce a su vez a concentraciones incrementadas de carbono orgánico en los sedimentos que se depositan de esta agua.
[64] El enterramiento de la pirita, en contraste, sólo suele estimularse bajo condiciones euxínicas de la columna de agua (cuando la columna de agua contiene sulfuro). En lo que sigue, me referiré a columnas de agua anóxicas en general, lo que puede significar cualquiera de los tipos que acabamos de ver, a menos que señale otra cosa.
Hecha esta digresión, podemos imaginar que una retroalimentación de oxígeno estuvo detrás de la expansión y contracción de las condiciones anóxicas del mar. Lo esperable, en general, es que el grado de anoxia en las aguas profundas del océano crezca cuando la concentración atmosférica de oxígeno se reduce. Esto tiene sentido porque, con menores niveles de oxígeno en la atmósfera, menos oxígeno será mezclado y transportado al océano profundo. Sin embargo, con el mar Negro como ejemplo, cuando las condiciones anóxicas se expanden, las tasas de enterramiento de carbono orgánico y sulfuros de la pirita (si la columna de agua es euxínica) deben incrementarse también. El enterramiento aumentado de carbono orgánico y pirita generaría una fuente mayor de oxígeno para la atmósfera y por tanto incrementaría la concentración de oxígeno atmosférico. Los niveles elevados de oxígeno, a su vez, reducirán el grado de anoxia en el océano; esto merma las tasas de enterramiento de carbono orgánico y pirita y por tanto reduce la fuerza de la fuente de oxígeno atmosférico. Esta cadena de acontecimientos aporta un bonito mecanismo de estabilización (retroalimentación negativa) para evitar que las concentraciones de oxígeno se eleven o reduzcan demasiado.
Si profundizamos más en el asunto, encontramos que la anoxia oceánica produce retroalimentaciones adicionales sobre la concentración de oxígeno. Para entenderlo tenemos que seguir el destino del nitrógeno y el fósforo, dos de los más importantes nutrientes cuya disponibilidad controla las tasas de producción primaria en los océanos. Empecemos con el fósforo. La mayoría de los geoquímicos, de hecho, suelen ver la disponibilidad de fósforo como el factor más importante para el control de las tasas de producción primaria en el océano.
[65] Con esto en mente, mi estudiante de doctorado en Yale Ellery Ingall, ahora en el Instituto de Tecnología de Georgia, en Atlanta, hizo un descubrimiento asombroso durante su investigación para la tesis. Encontró que tanto los sedimentos modernos como los primitivos depositados en contextos anóxicos, como el mar Negro, contienen mucho menos fósforo que los sedimentos depositados bajo columnas de agua que contienen oxígeno.
[66] Esta observación revela retroalimentaciones adicionales sobre la concentración atmosférica de oxígeno. Así, en vista de las observaciones de Ellery, unas condiciones anóxicas expansivas deben resultar en una menor retirada de fósforo desde la columna de agua hacia los sedimentos marinos. Ya sabemos que las condiciones anóxicas estimulan el enterramiento de carbono orgánico —y de pirita en algunos casos—, pero con una menor retirada de fósforo desde el agua hacia los sedimentos debería haber más fósforo disponible para avivar unas tasas de producción primaria aún mayores. Esto resultaría en aún mayores tasas de enterramiento de carbono orgánico y de liberación de oxígeno a la atmósfera. Por tanto, el ciclo del fósforo actúa reforzando la retroalimentación negativa que acabamos de discutir en relación con el papel de la anoxia en el mantenimiento de unos altos niveles de oxígeno.
Vemos el mismo efecto de refuerzo cuando los niveles de oxígeno aumentan. Cuando esto ocurre se reduce la extensión de agua oceánica anóxica. Esto se acompaña por un incremento en la extensión de las aguas oxigenadas, con el consiguiente aumento en la tasa de retirada de fósforo desde estas aguas oxigenadas hacia los sedimentos depositados. Esta retirada de fósforo más rápida disminuye la cantidad de fósforo en el agua oceánica, lo que debería reducir la cantidad de fósforo en el océano, que a su vez reduciría las tasas de producción primaria. Una tasa reducida de producción primaria conduciría a una tasa reducida de enterramiento de carbono orgánico y, por tanto, a una tasa reducida de liberación de oxígeno a la atmósfera. Esto evita que las concentraciones de oxígeno se hagan demasiado altas. Estas elegantes ideas fueron presentadas en primer lugar por Ellery, en colaboración con Philippe van Cappellen, otro de mis estudiantes de doctorado de la época, y trabajando con Bob Berner.
Toda esta discusión sobre el fósforo no debe hacer que nos olvidemos del nitrógeno. Ya echamos un vistazo al ciclo del nitrógeno en el último capítulo, cuando discutimos cómo los microbios anaeróbicos retiran el nitrato como gas N2 en las zonas oceánicas de mínimo oxígeno. También discutimos cómo las cianobacterias fijadoras de nitrógeno reconvierten el gas N2 en amonio, resuministrando nitrógeno a la biosfera. Se puede imaginar que, si la anoxia oceánica se extendiera, los procesos anaeróbicos que producen gas N2 ganarían importancia y podrían drenar del mar el nitrógeno biológicamente disponible (nitrato en su mayor parte). Por tanto, y en contraste de lo que ocurre con el fósforo, la extensión de la anoxia en los océanos globales tendería a limitar la disponibilidad de nitrógeno y, potencialmente, la producción primaria. El grado de esta limitación dependería crucialmente de si la fijación de nitrógeno pudiera acelerarse para reponer el nitrógeno perdido. Si la reposición por fijación de nitrógeno compensara exactamente la pérdida de nitrógeno como gas N2, el mecanismo de retroalimentación por fósforo descrito más arriba controlaría las tasas de producción primaria y de enterramiento de carbono orgánico. Sin embargo, si la fijación de nitrógeno no pudiera estar a la altura de la pérdida de nitrógeno, las concentraciones de nitrógeno se desplomarían y limitarían la producción primaria. Si fuera así, la retroalimentación por fósforo descrita arriba sería infructuosa.
Mi amigo y colega Paul Falkowski, de la Universidad de Rutgers, cree que este último sería el caso. En sus modelos, de hecho, la anoxia oceánica tiene el efecto de disminuir las tasas de producción primaria mediante una drástica limitación de nitrógeno. Si el nitrógeno verdaderamente limita la producción primaria en tiempos de anoxia oceánica extendida, el resultado es una retroalimentación positiva muy interesante. Para verlo, imagina que tenemos precisamente la situación descrita más arriba: una anoxia generalizada en el mar con una grave limitación de nitrógeno sobre la producción primaria. Ahora imaginemos que, por alguna razón, el nivel de oxígeno atmosférico sube sólo un poco. Esto haría disminuir la anoxia en los océanos y, siguiendo el argumento esbozado antes, reduciría las tasas de pérdida de N2 por desnitrificación. Puesto que menos nitrato es retirado del océano como gas N2, parte de la limitación de nitrógeno se vería aliviada, lo que conduciría a una tasa incrementada de producción primaria y enterramiento de carbono orgánico. Esto produciría más oxígeno, reduciendo aún más la extensión de la anoxia oceánica, y así sucesivamente. En principio, esta retroalimentación positiva seguiría actuando hasta que el nitrógeno dejara de ser el nutriente limitante. La retroalimentación por fósforo tomaría entonces el relevo para regular el oxígeno atmosférico.
Hay una última retroalimentación que debemos considerar. Los que trabajamos con bombonas de gas comprimido sabemos que el oxígeno puro es una sustancia muy reactiva. Mantenemos bien alejada de él cualquier chispa o llama, y sólo lo usamos cuando nos consta que hay buena ventilación, o mejor aún, en una campana extractora donde cualquier exceso de gas es retirado rápidamente de la habitación. Si tienes una bombona de oxígeno en casa acoplada a una mascarilla de respiración, ya conoces las normas: ni cigarrillos, ni chispas ni llamas de ninguna clase cerca del equipo de oxígeno. La razón básica para todas estas precauciones es que las cosas combustibles arden mucho mejor en oxígeno puro que en el aire. Los experimentos de combustión indican que, si duplicáramos la concentración de oxígeno atmosférico respecto a la actual, cosas como los árboles o la hierba arderían mucho más fácilmente. Bien, pues ahora viene la retroalimentación. La idea es que, si la concentración de oxígeno sube demasiado, las plantas terrestres las pasarán canutas para prosperar, porque arderán con facilidad a la mínima chispa que se produzca, por ejemplo en una tormenta. Esta idea surge de experimentos dirigidos por Andy Watson, actualmente en la Universidad de East Anglia, como parte de su tesis doctoral.
[67] Lo que implica es que unos altos niveles de oxígeno deberían causar fuegos forestales más desenfrenados y reducirían el crecimiento de las plantas en tierra firme. Esta retroalimentación por fuego, en consecuencia, puede ser importante para definir un nivel máximo de oxígeno en la atmósfera.
Por fortuna para nosotros, y para toda la vida de la Tierra, parece haber muchos procesos (retroalimentaciones positivas y negativas) que controlan y estabilizan la concentración de oxígeno en la atmósfera. Como veremos en capítulos subsiguientes, diferentes retroalimentaciones han actuado para controlar el oxígeno en distintos periodos de la historia de la Tierra. Aclarar estas retroalimentaciones nos permite apreciar plenamente la interacción entre la biología, la química y la geología para dar forma al entorno químico de la superficie de la Tierra, y cómo esta interacción ha cambiado con el tiempo.
Capítulo 6
La historia temprana del oxígeno atmosférico: evidencias biológicas
Se atribuye al filósofo francés del siglo XII Bernardo de Chartres la siguiente cita:
Somos como enanos subidos a hombros de gigantes, y así podemos ver más que ellos, y a mayor distancia, no por nuestra agudeza visual o algún otro mérito, sino porque nos levantan y crecemos hasta su enorme altura.[68]
Este sentimiento ha perdurado a través de los siglos, y es tan cierto hoy como hace 900 años. En este capítulo iniciamos la discusión de la historia del oxígeno atmosférico a través del tiempo geológico (véase la figura P.1 para la escala de tiempo geológica), y uno de los gigantes de esta discusión es Vladimir Vernadsky, el mineralogista ucranio convertido en geoquímico y pensador visionario. Vernadsky nació en 1863 y murió en 1945. Su vida transcurrió, por tanto, a través de una increíble agitación, incluyendo dos guerras mundiales y la caída de la Rusia zarista. En 1926 publicó su obra magna, La biosfera, donde exploró sistemáticamente el modo en que la vida funciona como una fuerza geológica. En este libro sus observaciones fueron agudas, profundas sus reflexiones, grandes sus pronunciamientos. Uno de los asuntos que trató fue la historia del oxígeno atmosférico. Así empezaba su discusión:
En todos los periodos geológicos, la influencia química de la materia viva sobre su entorno no ha cambiado de manera significativa; los mismos procesos de erosión han funcionado sobre la superficie de la Tierra durante todo ese tiempo, y la composición química media tanto de la materia viva como de la corteza terrestre han sido más o menos iguales a las de hoy.
Por tanto, mirando a las rocas a través del tiempo, por todo lo que sabía Vernadsky, la influencia de la vida en la superficie de la Tierra había permanecido aproximadamente igual. Esto le condujo a concluir, un poco más tarde en el texto:
El fenómeno de la erosión superficial muestra claramente que el oxígeno libre tuvo el mismo papel en el eón Arcaico que en la actualidad… El dominio de los fotosintetizadotes en aquellos tiempos distantes fue la fuente del oxígeno libre, casi todo el cual fue del mismo orden que hoy.
Cuando leí estos pasajes por primera vez, me dejaron pensando. Se trata de la primera explicación, que yo sepa, de la historia del oxígeno atmosférico a través del tiempo. No es muy detallada, desde luego, y cuando prosigamos con nuestra discusión puede que no coincidamos con la conclusión de Vernadsky. Pero eso no es lo importante. Lo importante es que él imaginó que la historia del oxígeno atmosférico era un problema científico tratable, determinó qué evidencia podría usarse para abordarlo, y lo hizo dentro de los límites de las observaciones disponibles en la época. Impresionante, en mi opinión.
Aunque Vernadsky es un héroe científico en Rusia, por desgracia apenas se le conoce en Occidente.
[69] Parte del problema es que la primera traducción al inglés de
La biosfera no se publicó hasta 1977, y quizá peor aún, el telón de acero y la guerra fría que siguieron a la segunda guerra mundial limitaron notablemente el intercambio científico entre los países del bloque soviético y Occidente. Por lo que a mi respecta, oí hablar por primera vez de Vernadsky hace unos 10 años, y no leí
La biosfera hasta hace muy poco. Me resultó evidente al leerla, sin embargo, que Vernadsky hablaba un lenguaje que yo podía entender. De hecho, muchas de sus ideas encajan bien en nuestra comprensión actual del acoplamiento entre la biosfera y la geosfera.
¿Cómo es que Vernadsky suena tan familiar si había pasado inadvertido para la ciencia occidental? Una explicación es que, cuando las ideas científicas son realmente buenas, ofrecen un entendimiento extraordinario del funcionamiento del mundo natural. Por tanto, la ciencia convergerá en esas ideas, si no ahora por un científico, más tarde por otro. Ya hemos visto un ejemplo de esto en el capítulo 5, donde Jacques Joseph Ebelmen describió correctamente en 1845 los mecanismos geológicos que controlan la concentración de oxígeno atmosférico. Estas ideas se perdieron y fueron redescubiertas independientemente por Bob Garrels, Ed Perry y Dick Holland unos 130 años después.
[70] Así, es posible que hayamos redescubierto en Occidente mucho de lo que Vernadsky entendió hace décadas.
También es posible que las ideas de Vernadsky nos hayan llegado por rutas indirectas. En los años cincuenta y sesenta los científicos rusos estaban kilómetros por delante de los occidentales en el entendimiento del papel de los microbios en la química de las aguas naturales y los sedimentos. Todavía no he podido confirmarlo, pero dado el estatus de Vernadsky en la ciencia rusa, tiene que haber ejercido una fuerte influencia en los ecólogos microbianos de ese país. Aunque gran parte de su trabajo se publicó en ruso y no llegó a los científicos occidentales, hubo varios encuentros importantes entre científicos rusos y occidentales en los años setenta y los ochenta, y las conclusiones de esas conferencias se presentaron en diversos volúmenes influyentes.
[71] Es posible que Vernadsky, al menos en parte, llegara a nosotros por esa vía.
Volvamos ahora, en cualquier caso, a las reflexiones de Vernadsky, y planteémonos específicamente la siguiente cuestión: ¿cómo podemos saber algo sobre la historia del oxígeno atmosférico? Por supuesto, necesitamos pistas. Vernadsky las halló en las rocas sedimentarias antiguas que antaño formaron parte del fondo marino primitivo. Eso ha resultado ser una idea excelente, pero sería justo preguntarse cómo es que esos barros antiguos contienen pistas sobre los niveles de oxígeno atmosférico en el pasado. Echemos un vistazo.
Si alguna vez has caminado descalzo por la arena húmeda de la playa, habrás sentido que el barro se desliza entre los dedos de tus pies, y tal vez te hayas fijado en que se forman burbujas cuando los pies se hunden en el barro. Las burbujas son de metano, formadas por los mismos microbios metanógenos que vimos en el capítulo 2. En este caso, se alimentan de la materia orgánica del fango. La presencia de metanógenos nos dice algo sobre la química del barro y la ecología de los microbios que viven allí. Es posible que hayas notado un tufillo a sulfuro. Este gas es producido por otro grupo de microbios, los mismos reductores de sulfato que vimos en capítulos anteriores, al alimentarse de residuos orgánicos. Otra pista. Notas una cosa dura entre los dedos de los pies. Te agachas para cogerla y ves que es una concha vacía. Otra pista más. La materia orgánica del fango también contiene pistas. Proviene de organismos muertos que pueden estar en el agua que cubre el fango o en el fango mismo. Con suerte, esta materia orgánica puede decirnos algo sobre los organismos que vivían en el entorno. Las propias partículas del fango reaccionan con componentes químicos del agua marina mientras sedimentan sobre el fondo. El fango sigue reaccionando cuando ya se ha asentado en el fondo, y mientras se van acumulando el sulfuro y el metano. Más pistas aún. En conjunto, el fango está repleto de pistas. Nuestro desafío, como veremos a medida que la historia se desarrolle, es percibir qué pistas hay allí y cómo se relacionan con el contenido de oxígeno de la atmósfera. De modo que Vernadsky estaba en lo cierto, las pistas están en el barro, pero entenderlas no siempre resulta fácil.
Bien, tomemos un manojo de rocas sedimentarias y empecemos. Podemos acercarnos a la tienda de rocas y pedir a la dependienta unas rocas de, pongamos, las zonas someras y profundas del océano (¿por qué no?, compararlas puede ser interesante). Tomemos muestras de cada 10 millones de años en diez lugares alrededor del mundo (queremos asegurarnos una buena cobertura) desde el nacimiento del planeta 4550 millones de años atrás hasta 2500 millones de años atrás (el final del eón Arcaico, y el foco del presente capítulo). Eso es un montón de rocas, pero qué demonios, más vale que sobren que no que falten. La dependienta pone cara de incredulidad y abre la puerta del almacén de par en par. De inmediato nos damos cuenta del problema. Las estanterías están casi vacías. Por mucho que la empleada quiera ayudar, las rocas simplemente no están ahí. De hecho, no hay nada en las estanterías salvo unos cuantos granitos de mineral de hace más de 4000 millones de años.
[72] Los primeros 500 millones años de la historia de la Tierra no están representados en el registro geológico. Encontramos unas pocas rocas de hace 3800 millones de años, algunas más de 3500, algunas de 3200, 3000, 2900, 2700 y algunas incluso más jóvenes, con una cobertura bastante mejor hasta 2500 millones de años atrás. Pero, en general, el registro geológico primitivo es muy pobre. El problema es que los mismos procesos tectónicos que discutimos en el capítulo 1 —la subducción, la construcción de montañas, la erosión y el reciclado constante de rocas en la superficie de la Tierra—, todos esos procesos que ayudan a hacer de la Tierra un planeta magníficamente habitable, también causan estragos en el registro geológico. Y cuanto más viejas sean las rocas, más probable es que se hayan perdido por erosión o que hayan sido engullidas y transformadas por las maceraciones de la Tierra.
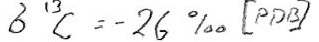
FIGURA 6.1 Esta fórmula está escrita con el grafito de unas rocas sedimentarias metamórficas de Isua, Groenlandia. El texto es un homenaje a la típica composición isotópica del carbono orgánico en los sedimentos. Foto cortesía de Minik Rosing.
Le damos las gracias a la mujer de la tienda de rocas, tomamos lo que podemos y nos vamos de allí. Nos paramos a la sombra de un árbol cercano y vemos lo que tenemos. Al mirarlas más detenidamente resulta que las rocas más viejas no sólo son escasas, sino que en muy mala forma. Quiero decir que muchas de ellas han sido calentadas a altas temperaturas, y a veces más de una vez. Este calentamiento cambia los minerales originales de la roca a otras formas, por el proceso llamado «metamorfismo». Por poner un ejemplo bastante revelador, la materia orgánica original de algunas de nuestras muestras más antiguas, las rocas de 3800 millones de años de Isua, en Groenlandia, se ha convertido por completo en grafito debido al calor. Uno puede, de hecho, escribir con estas rocas (figura 6.1). En las rocas de Groenlandia, y en muchas otras, ocurre también que los líquidos han fluido a través de ellas cuando estaban profundamente enterradas en la corteza terrestre, a veces redistribuyendo los constituyentes químicos.
[73] Mi intención aquí no es resultar desalentador, sino más bien ser realista sobre las limitaciones del registro geológico, sobre todo cuando queremos mirar muy atrás en el tiempo. Con todo, a pesar de las dificultades, hay algunas cosas que podemos decir tanto de la química como de la biología de la Tierra más primitiva. Empezaremos explorando la biología arcaica. En este capítulo nos interesará en especial buscar cualquier signo de cianobacterias. Recordemos que fueron las primeras productoras de oxígeno en la Tierra, y que fueron las estrellas del capítulo 4. Sin cianobacterias, el oxígeno no habría podido acumular en la atmósfera. En el próximo capítulo nos centraremos en la evidencia química de oxígeno en la Tierra primitiva.
Empecemos nuestra expedición por las rocas ricas en grafito de 3800 millones de años de antigüedad que hallamos en Isua, Groenlandia. Mi amigo y colega Minik Rosing, del Museo Geológico de Dinamarca (a quien encontramos brevemente en el capítulo 2) ha pasado gran parte de su carrera investigando las rocas de Groenlandia. La verdad es que Groenlandia es un lugar especial para Minik, puesto que nació allí, y su padre, Jens Rosing, era un conocido pintor, ilustrador, diseñador de joyas y escritor en aquel país. Tras muchos años de trabajo de campo, Minik descubrió esas rocas sedimentarias ricas en grafito y de inmediato las reconoció como buenas candidatas para revelar la naturaleza de la vida primitiva en la Tierra. Los sedimentos en sí mismos, expuestos en un afloramiento no mucho mayor que un coche, se depositaron aparentemente en aguas marinas profundas. Las capas ricas en grafito aparecen entreveradas con capas de sedimento conocidas como turbiditas.
[74] Estas se forman como sedimentos en aguas más someras, pero se removilizan y se transportan deprisa pendiente abajo y acaban depositadas en aguas profundas (figura 6.2). Minik interpreta las capas de grafito como deposiciones continuas de partículas ricas en materia orgánica que provienen de las aguas superficiales del océano. Este flujo constante y suave de materia orgánica fue puntuado por el ocasional flujo rápido de turbiditas. De esta forma se obtiene una alternancia, como en una tarta de galletas, entre los sedimentos orgánicos y las turbiditas. El flujo de materia orgánica a los sedimentos debió ser razonablemente alto para generar el suficiente grafito como para que se pueda escribir con él. ¿Pero de qué forma de vida provenía esa materia orgánica? Esta es la pregunta del millón. ¿Pudo haber sido producida por cianobacterias?

FIGURA 6.2 Rocas sedimentarias de Isua, Groenlandia. Nótese la capa negra y casi vertical rica en grafito que va desde arriba (centro) hasta abajo (izquierda). Hay otra capa similar a la derecha. Foto cortesía de Minik Rosing.
Los sedimentos están demasiado cocinados para ofrecer algún tipo de evidencia fósil sobre el tipo de organismos que estaban presentes, pero hay otros indicios que podemos utilizar. Toma una roca, gírala en tu mano y mira largo y tendido al grafito. ¿Podemos encontrar ahí algunas pistas?
[75] Minik hizo esto y decidió buscar evidencias en los isótopos de carbono preservados en el grafito. Para comprender esto, recordemos que el carbono se presenta en la naturaleza como tres diferentes isótopos: carbono-12, carbono-13 y carbono-14. Mucha gente ha oído hablar del carbono-14; es radiactivo, se forma en la atmósfera y aguanta sólo unas decenas de miles de años, así que no nos ocuparemos más de él. El carbono-12 tiene seis protones y seis neutrones, mientras que el carbono-13 tiene seis protones y siete neutrones. Por tanto, el carbono-13 es cerca de un 8% más pesado que el carbono-12 (13/12 = 1.083). Químicamente, los dos isótopos de carbono son casi idénticos, pero no del todo, y como veremos estas pequeñas diferencias en reactividad química dan lugar a señales que podemos detectar e interpretar.
Volvamos ahora a la cuestión de la vida. Como exploramos en los capítulos 2 y 3, muchos organismos fabrican sus células con dióxido de carbono (CO
2) derivado de la atmósfera o disuelto en agua (las plantas, por ejemplo, hacen esto mediante la fotosíntesis). Los organismos que hacen esto deben convertir el CO
2 en materia orgánica, y esto se logra con varios tipos diferentes de reacciones bioquímicas catalizadas por enzimas. Las plantas, como las cianobacterias, utilizan la enzima Rubisco. Ya hablamos de Rubisco en el capítulo 3, pero lo que no dijimos es que Rubisco usa preferentemente el carbono-12 del CO
2 sobre el carbono-13. Esto implica que las plantas y las cianobacterias están mermadas en carbono-13 (y al mismo tiempo enriquecidas en carbono-12) en comparación con la distribución de átomos de carbono-12 y carbono-13 en el CO
2 a partir del cual se formó la materia orgánica. Ahora necesitamos un lenguaje para expresar esas proporciones. Las proporciones de isótopos se suelen medir con unos instrumentos llamados espectrómetros de masas. La proporción de isótopos que medimos en nuestra muestra se expresa con relación a la proporción de un estándar con una ratio conocida de C-13 a C-12. El resultado es un número muy pequeño, así que lo multiplicamos por 1000 para obtener números fáciles de discutir. Para el sistema de carbono, expresamos la composición isotópica de nuestra muestra como δ
13C.
[76] Cuanto más positivo sea el valor de δ
13C, más enriquecida estará la muestra en C-13. Volviendo a Rubisco, esta enzima selecciona el C-12 con una preferencia del 2,5% en el CO
2 de la atmósfera. Si usamos la nomenclatura que acabamos de aprender, esto significa que la materia orgánica formada por Rubisco es 25 por mil mermada en C-13 respecto a la del CO
2.
[77]Minik midió el δ
13C del grafito de Isua y encontró que el grafito estaba mermado en C-13 por un 17 por mil (la diferencia isotópica entre el grafito y el carbono inorgánico de otras rocas sedimentarias en Isua), que es una magnitud coherente con su producción por cianobacterias (fig 6.3). Entonces, ¿tenemos una evidencia sólida de cianobacterias tan antiguas como 3800 millones de años atrás? Por desgracia, no. El problema es que muchos tipos de organismos, además de las cianobacterias, usan Rubisco y producen una señal similar de isótopos de carbono. De modo que no podemos concluir que las cianobacterias estuvieran presentes en Isua. Lo que sí tenemos es una evidencia bastante buena de que la vida estaba allí. La señal isotópica de carbono de Isua es coherente con la presencia de vida, como lo es el escenario geológico. La materia orgánica sedimentó desde las aguas superficiales del océano, como cabría esperar de organismos que fijaran CO
2 en el océano. Verdaderamente, si no fuera de la vida, resulta muy difícil imaginar de dónde habría venido la materia orgánica.
[78] Aunque no demos detalles sobre los organismos específicos, tenemos una evidencia bastante buena de que en los tiempos de Isua, alguna forma de vida estaba haciendo materia orgánica a partir de CO
2 en las capas superficiales del océano.
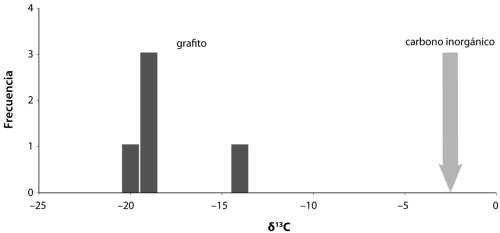
FIGURA 6.3 Composición isotópica del grafito y el carbono inorgánico de las rocas sedimentarias de Isua (la distribución de frecuencia de los análisis sólo se aplica al grafito).
¿Podemos dirigir la atención a alguna otra parte en nuestra búsqueda de cianobacterias? ¿Qué tal los fósiles? ¿Vemos algo en los fósiles más antiguos que se parezca a las cianobacterias? Pensábamos que sí. Bill Schopf, de la Universidad de California en Los Ángeles (UCLA), es famoso por haber descrito estructuras parecidas a fósiles en el Apex Chert de Australia occidental, datadas en 3500 millones de años. La figura 6.4 muestra algunos ejemplos. Aunque no están terriblemente bien conservadas, muchas de estas estructuras parecen consistir de múltiples células organizadas en filamentos (los llamados tricomas), y los filamentos son grandes (para los estándares microbianos), llegando hasta las 50 micras (0,05 milímetros) de longitud. Bill ha hecho varias compilaciones de los tamaños de diversos grupos microbianos, y las cianobacterias tienden a ser mayores que los demás. De hecho, dominan en un intervalo de tamaños que cuadran con los observados en estos fósiles del Apex. Reuniendo esta información, Bill concluyó que estos fósiles representan «probables» cianobacterias; aunque no suponían una prueba de cianobacterias, los fósiles se tomaron por una evidencia bastante convincente. Digo que no suponen una prueba por al menos dos razones. La primera es que los fósiles no están bien preservados y, aparte del tamaño, no se observó ninguna característica convincente de las cianobacterias.
[79] Otro problema, también conocido ya en la época, es que algunos organismos no cianobacterianos producen filamentos de tamaño y forma similares a los de Apex Chert.
[80] Sin embargo, pese a estas prevenciones, muchos científicos (quizá incluso la mayoría) sostuvieron la opinión de que estos fósiles de Apex Chert representaban «probables» cianobacterias, y esta idea perduró por una década.
Hace unos 10 años, Martin Brasier, de la Universidad de Oxford, reexaminó muchos de los fósiles de Bill, que se encuentran en finas secciones de roca hechas especialmente para observarse al microscopio óptico. Con unas técnicas mejoradas de imagen tridimensional, Martin pudo reconocer unas ramificaciones asociadas con algunos de los fósiles originales de Bill, que no habían sido apreciadas por Bill en sus descripciones. El tipo de ramificaciones observadas por Martin no parecía tener correspondencias obvias en el mundo microbiano. Martin también examinó las rocas cuidadosamente en busca de otros posibles rasgos biogénicos y encontró que había mucho carbono orgánico en la roca, pero variaba en tamaño desde gotitas informes hasta estructuras más organizadas semejantes a fósiles. Al final, Martin concluyó, al contrario que Bill, que los rasgos interpretados originalmente como «cianobacterias» no eran cianobacterias en absoluto, y de hecho ni siquiera eran fósiles. Según Martin, esos pseudofósiles se explicaban mejor como el resultado de procesos inorgánicos. Explicó que la materia orgánica, móvil en esas rocas cuando se calentaban a las altas temperaturas de las profundidades de la Tierra, se concentraba alrededor de granos de cuarzo dando lugar a diversas formas e incluso a acumulaciones parecidas a fósiles en algunos casos. Martin también señaló que la geología de los depósitos puede ser más compleja de lo que se había apreciado originalmente. Mientras que Bill había considerado que esos depósitos se habían acumulado en una playa o algo similar, o en la desembocadura de un río, las reconstrucciones más recientes sugieren que se formaron en las profundidades de la Tierra. Un escenario de esa profundidad no sería el lugar esperable para las cianobacterias, dada su necesidad de luz, a menos que los trozos de roca que contienen los fósiles hubieran sido transportados de alguna manera desde un entorno más favorable.
FIGURA 6.4 Fósiles del pedernal de Apex. De Schopf y Kudryavtsev (2012). Reproducido con permiso.
Como en una partida de póquer de grandes apuestas, las preocupaciones de Martin forzaron una respuesta de Bill, en la que utilizó unas técnicas de microscopía aún más avanzadas (espectroscopia Raman) para verificar que las paredes de los «fósiles» eran en realidad querógeno (un tipo de materia orgánica resistente), y que el querógeno parecía formar, al menos en algunos casos, compartimentos discretos similares a células (véase la figura 6.5). Martin volvió a la carga y arguyó que esas «células» eran meramente carbono orgánico que revestía granos de cuarzo. Bill volvió a replicar que no era así. Es probable que todavía no hayamos visto el final de esta discusión, pero la idea original de que esas formas representan «probables» cianobacterias ha sido barrida del mapa, incluso por Bill. Por tanto, si el argumento se centra en si esas formas representan evidencias de vida primitiva en la Tierra, las evidencias pueden estar ahí, pero hay otras pruebas que son más antiguas y menos polémicas. De hecho, hay buenas evidencias de vida en los isótopos de carbono preservados en las más viejas rocas de Isua, como vimos antes. Más aún, los isótopos de carbono en rocas de una edad similar a las del Apex Chert parecen aportar también evidencias de vida.
[81] Y, como exploramos en el capítulo 2, también hay evidencias de metabolismos microbianos específicos como la reducción de sulfato y la metanogénesis por esa misma época.
Así que otra percepción potencialmente prometedora de la antigüedad de las cianobacterias ha mordido el polvo. ¿Y ahora qué? ¿Hay alguna otra cosa a la que podamos mirar? Hay, de hecho, otros tipos de fósiles que podrían ayudar. Cuando pensamos en fósiles solemos imaginar huesos o conchas o, en el caso de los microbios, las propias células. Sin embargo, las células, y en particular las membranas celulares, también contienen compuestos que se pueden preservar en las rocas. Las estructuras celulares pueden haberse perdido, pero algunos de sus componentes químicos pueden perdurar en formas químicas conocidas como biomarcadores. Algunos de estos biomarcadores son distintivos de ciertos tipos de organismos. Esto suena bastante prometedor, pero hay algunos asuntos significativos que debemos considerar. Un problema es que en rocas realmente viejas, como las que estamos discutiendo ahora, sólo podemos esperar hallar cantidades minúsculas de moléculas biomarcadoras. Incluso en los mejores casos, estas rocas se han calentado lo bastante como para transformar la mayoría de los biomarcadores reconocibles en formas irreconocibles. Otro problema, cuya importancia se duplica por las bajas cantidades de biomarcadores en la roca, es la contaminación por materiales más recientes, y esta contaminación puede venir de casi cualquier parte.

FIGURA 6.5 Uno de los fósiles del pedernal de Apex, mostrado en una imagen de microscopio óptico arriba a la izquierda (a). El panel (c) muestra una imagen Raman 3D de la caja rectangular del panel (a). Los paneles (d-h) muestran imágenes Raman 2D en una serie de campos focales. La imagen de arriba a la derecha (b) muestra los fósiles fuera del plano focal, pero subraya que los fósiles no están formados alrededor de granos de cuarzo, señalados por las flechas. De Schopf y Kudryavtsev (2012). Reproducido con permiso.
Las mejores rocas para este tipo de estudio se obtienen taladrando en busca de material fresco enterrado bajo la superficie de la Tierra. Pero el proceso habitual de taladrado suele utilizar lubricantes de petróleo. ¡Hablando de contaminación! De modo que el taladrado ha de hacerse con cuidado y en ausencia de sustancias orgánicas. La aventura entera, sin embargo, puede ser una causa perdida desde el principio si fluidos ricos en sustancias orgánicas han migrado a través de las rocas mientras todavía estaban bajo tierra, y esto podría haber ocurrido millones, o incluso miles de millones de años tras la deposición inicial. Basta pensar en cómo el petróleo migra para formar reservorios bajo la superficie. Otro problema es el manejo. Incluso cuando nos hemos convencido de que hemos recogido muestras incontaminadas, debemos tener cuidado para no introducir contaminación cuando almacenamos y procesamos las muestras, y esto es más difícil de lo que parece. Folículos pilosos sueltos, polvo, polen, huellas digitales o una miríada de compuestos repugnantes derivados del lugar donde se almacenan las muestras; todos ellos son fuentes de contaminación. No es un trabajo para torpes, ni para pusilánimes.
Roger Summons, del MIT en Boston, se preocupa incesantemente sobre todos estos problemas, y tiene uno de los pocos laboratorios en el mundo que estudia biomarcadores en rocas realmente antiguas. Su reciente y muy dotado estudiante de doctorado Jake Waldbauer, ahora en la Universidad de Chicago, ha trabajado en rocas entre 2460 y 2670 millones de años de antigüedad procedentes de Sudáfrica. Las rocas fueron taladradas con cuidado, recogidas con cuidado y manejadas con cuidado. Las rocas en sí mismas carecían de cualquier migración obvia de hidrocarburos o generación de hidrocarburos, y tras la extracción las pequeñas cantidades de biomarcadores recuperadas tenían las características de la materia orgánica que ha sido parcialmente degradada por calentamiento. Esto significa que no hay ninguna evidencia de contaminación reciente. En suma, Jake, Roger y sus colegas han hecho el mejor trabajo posible.
Lo que han encontrado es extremadamente interesante. Descubrieron varios tipos diferentes de moléculas biomarcadoras, y lo que más nos interesa ahora es que también encontraron diversas moléculas de esterano.
[82] Los esteranos son derivados de los esteroles, de los que el colesterol es el ejemplo más conocido. Los esteroles se concentran en la membrana celular, donde contribuyen a incrementar la fluidez y flexibilidad de la membrana. De hecho, los esteroles se distribuyen de forma universal entre los organismos eucariotas,
[83] incluidas las plantas, los animales y los hongos. De manera crítica, por todo lo que sabemos, los esteroles requieren oxígeno para sintetizarse en la célula. Por tanto, allí donde encuentras esteroles, es que hubo oxígeno libre, aunque no necesariamente muchísimo. En otro trabajo, Jake mostró que los esteroles son sintetizados por la levadura (sí, la levadura también es eucariota) con niveles de oxígeno unas 100 000 veces menores de los que uno encuentra hoy en agua saturada con aire. Por tanto, en una línea de razonamiento inteligente aunque algo indirecta, la presencia de esteroles significa presencia de oxígeno y, en consecuencia, presencia de cianobacterias. Parece, entonces, que Jake (junto a Roger y otros implicados en el estudio) ha encontrado evidencia de producción de oxígeno por cianobacterias tan pronto como 2670 millones de años atrás, si no antes. Al fin, esto es algo de lo que podemos fiarnos. Bueno, probablemente: el pelo en la sopa en este caso es si, pese a su gran meticulosidad, las rocas de Jake siguen estando contaminadas. Jochen Brochs, un antiguo estudiante de Roger que ahora trabaja en la Universidad Nacional de Australia en Canberra, sostiene que probablemente lo están. El tiempo lo dirá, con seguridad.
Esta búsqueda de cianobacterias ha tenido alturas vertiginosas y bajuras deprimentes, y acaba con un brochazo de realismo. El problema es difícil. Las rocas no están en buena forma, y no hay muchas. Así está el tema. Ello no significa, sin embargo, que la gente vaya a dejar de investigar. Tal vez ocurra un gran descubrimiento, quizás unas cuantas células cianobacterianas de 3500 millones de años maravillosamente preservadas esperen en alguna parte bajo tierra, hasta ahora fuera de nuestro alcance, y queden expuestas por casualidad o por el empeño de algún científico curioso en el futuro. O tal vez se desarrolle algún otro método ingenioso para buscar cianobacterias. De hecho, esto es en parte de lo que trata el próximo capítulo. Exploraremos la química de la Tierra primitiva y trataremos de usar estrategias químicas para descubrir cualquier signo de oxígeno.
Capítulo 7
La historia antigua del oxígeno atmosférico: evidencias geológicas
Cuando yo era un estudiante de doctorado y estaba justo interesándome por la historia del oxígeno atmosférico, parecía que desenredar esa historia era una tarea de aficionados y soñadores, no para que los científicos serios perdieran el tiempo. Parecía que cualquiera con una idea extravagante podía entrar en ese campo, soltar su idea y largarse de inmediato. Las restricciones eran mínimas, de modo que hasta la idea más chiflada lograba alguna audiencia.
Estoy exagerando, por supuesto. Había unos pocos científicos muy serios desesperados por revelar la historia del oxígeno atmosférico en la Tierra primitiva, y uno de ellos era Dick Holland, de la Universidad de Harvard.
[84] Leí los trabajos de Dick casi inmediatamente tras empezar la tesis, y me dio una lección de humildad por su habilidad para orientarse a través de datos complejos y problemas complejos, y para extraer de allí principios simples (mi mentor de tesis Bob Berner, a quien conoceremos más a fondo en el capítulo 11, comparte ese talento). Esto requiere un sentido para el reconocimiento de patrones y el razonamiento lateral que la mayoría de nosotros, simplemente, no poseemos. Pero Dick lo tenía a espuertas. Cuando Dick terminaba de dar una charla, tú decías: «Sí, por supuesto, pero ¿por qué no se me ocurrió eso a mi?». Pero el caso es que no se te ocurrió a ti, ni a ningún otro.
Dick siguió muchas líneas de investigación durante su larga carrera, pero intentar comprender la historia del oxígeno atmosférico fue una constante a través de todo ello. Ya en 1962, Dick escribió un artículo extraordinario titulado
Un modelo para la evolución de la atmósfera de la Tierra. Empezaba con una clásica finura: «Subsiste cierto grado de incertidumbre sobre la composición de la atmósfera entre su origen y el paleozoico tardío».
[85] Más abajo resumía el verdadero estado de la cuestión: «Disponemos por tanto de sólo escasas evidencias sobre la química de la atmósfera durante gran parte del tiempo geológico».
En ese artículo, Holland se aproximaba a la historia del oxígeno atmosférico desde un ángulo muy diferente del que usó Vladimir Vernadsky. Vernadsky, a quien visitamos en el último capítulo, tenía un punto de vista uniforme y basado en su observación de que los sedimentos antiguos eran extraordinariamente similares a los que se están formando hoy. Muchas nuevas observaciones, sin embargo, se han acumulado desde tiempos de Vernadsky, poniendo en duda la probabilidad de que los niveles de oxígeno atmosférico hayan sido constantes durante el tiempo geológico, y el artículo de Dick de 1962 era un primer intento de esbozar una historia dinámica de los niveles de oxígeno atmosférico. Este trabajo es una clásica contribución de Dick Holland en muchos aspectos. En él, Dick ensambló con perspicacia toda la evidencia pertinente para discutir la historia del oxígeno atmosférico; luego organizó esta evidencia en un cuadro coherente, e intentó cuantificar las concentraciones pasadas de oxígeno mediante el razonamiento inteligente y el modelado cuidadoso. Otro distintivo del estilo de Holland es el libre reconocimiento de los problemas que existen y una discusión de las investigaciones futuras que pueden resultar fructíferas. De hecho, Dick vio el problema con tanta claridad que muchas de sus observaciones, y gran parte de su discusión, son aún hoy pertinentes y aportan un marco para la exposición que sigue.
Nuestro objetivo en este capítulo es explorar las evidencias geológicas y químicas sobre la historia del oxígeno atmosférico en la Tierra primitiva, con especial atención al eón Arcaico. Empezamos investigando algunos de los indicios que Dick discutió. Para hacer esto, nos colocamos bien nuestros cascos y penetramos hasta el fondo de las minas de oro de Sudáfrica. Algunas de estas minas, localizadas en la cuenca de Witwatersrand, cerca de Johannesburgo, han sido excavadas hasta profundidades de 3,9 kilómetros. Sin refrigeración, la temperatura en la mina subiría hasta unos abrasadores 55 ºC, pero con la maquinaria refrigeradora en funcionamiento podemos descender con seguridad y echar un vistazo a las rocas. Estas son las minas de oro más ricas del mundo, y se estima que han producido el 40% de todo el oro extraído en la historia. Si miramos con atención a las rocas, veremos que representan un antiguo depósito fluvial datado en 2800 a 3100 millones de años atrás. El oro en estos depósitos fue transportado por las fuertes corrientes fluviales y resultó atrapado ocasionalmente entre los cantos y arenas que formaban el lecho del río.
[86] Todo esto es muy similar a los depósitos fluviales excavados hace 150 años durante la gran fiebre del oro en California.

FIGURA 7.1 Imágenes por microscopio electrónico de barrido de piritas residuales (c y d), uraninita (f) y cromita (e) de los depósitos aluviales de Witwatersrand, Sudáfrica. La cromita (FeCr2O4) es también sensible al oxígeno, aunque la oxidación de la cromita parece requerir la influencia de microbios y oxidantes intermediarios como los óxidos de manganeso. Hoy se pueden encontrar cromitas residuales en los ríos. Imagen de Utter (1980), reproducida con permiso.
Nuestro guía nos ha suministrado un contador Geiger, y nos ha dicho que aquí no sólo hay oro para buscar.
Exploramos entre las piedras y de vez en cuando escuchamos el trino revelador del Geiger. Buscamos al culpable con nuestra lupa y vemos un grano esférico, ahora cementado por el cuarzo que lo rodea; pero en forma y carácter se parece mucho a los otros granos que hay a su alrededor (figura 7.1). Se nos dice que es uraninita, un mineral de óxido de uranio (UO2). El hecho de que este trozo de uraninita se haya desgastado hasta adoptar una forma esférica nos dice que fue transportado río abajo: saltando, rebotando y puliéndose en la corriente de agua, de modo similar al resto de las arenas y cantos que encontramos en este antiguo lecho de un río.
La uraninita, sin embargo, no es del todo igual a las otras piezas del sedimento fluvial. No la encontramos hoy en los ríos, y eso es porque reacciona fácilmente con el oxígeno, formando el ión uranilo soluble en agua (UO22+). Holland se dio cuenta de esto en su artículo de 1962, y utilizó la evidencia de las uraninitas de Witwatersrand para argumentar que había sólo «trazas» de oxígeno en la atmósfera cuando esos antiguos sedimentos fluviales se depositaron. Subsiguientemente se ha desarrollado un encendido debate que cuestiona la idea de que las uraninitas de Witwatersrand fueran en verdad cantos y arenas de río. Algunos han argumentado que el origen de las uraninitas hay que buscarlo en fluidos hidrotermales que más tarde circularon a través de las rocas, y que por lo tanto su presencia no dice nada sobre los antiguos niveles de oxígeno. Muchos otros, sin embargo, siguen apoyando la interpretación original de que la uraninita, o al menos parte de ella, forma parte de un antiguo depósito fluvial. La forma de los granos apoya este argumento y, cuando se ha datado, la uraninita parece ser más vieja que los sedimentos fluviales que la rodean. Esto sugiere que la uraninita fue liberada de rocas más antiguas durante la erosión y transportada río abajo, como se supuso inicialmente.
Felizmente, las uraninitas de origen fluvial no están restringidas a los antiguos depósitos de Witwatersrand en Sudáfrica. Birger Rasmussen, de la Universidad de Western Australia, y Roger Buick, ahora en la Universidad de Washington, han descrito uraninitas de río similares en sedimentos del oeste de Australia con edades que oscilan entre los 3250 y los 2750 millones de años. Asociados con estas uraninitas se hallan granos redondeados de pirita (FeS2, que vimos por primera vez en el capítulo 5) y también a veces otro mineral llamado siderita (FeCO3). La pirita es también muy abundante en los depósitos de Witwatersrand y, al igual que la uraninita, tanto la pirita como la siderita son sensibles al oxígeno. Tú mismo puedes intentar el experimento. Casi todas las tiendas de minerales tienen pirita. Compra un trozo barato, ponlo a la intemperie, espera y mira lo que pasa.
Otros antiguos depósitos fluviales de Canadá (de los que trataremos más en el próximo capítulo) e India muestran también signos de uraninita y pirita transportadas por ríos. En resumen, disponemos de evidencias de transporte de uraninita y pirita en ríos antiguos con edades que van de los 3250 a los 2450 millones de años. Lo que viene después de esa fecha es el tema del siguiente capítulo, pero la evidencia acumulada apoya la propuesta de Holland de que, como mucho, sólo había «trazas» de oxígeno en la atmósfera durante el eón Arcaico.
La presencia de minerales sensibles al oxígeno en antiguos depósitos fluviales supone una evidencia bastante convincente de bajas concentraciones de oxígeno en la atmósfera primitiva de la Tierra, pero el argumento se podría reforzar reuniendo más pruebas. Para encontrarlas, giremos la mirada de la tierra al mar. En el tiempo en que la uraninita, la pirita y la siderita eran transportadas como arenas de río, los océanos tenían una química muy diferente a la de hoy. Puede que el lector recuerde que en el capítulo 2 discutimos la deposición de un tipo de roca llamado formación de hierro bandeado (o a menudo BIF, por banded iron formation) en los océanos primitivos como evidencia de una química oceánica diferente de la actual. No dimos muchos detalles, sin embargo.
Nuestro objetivo ahora es mirar con más atención a estas formaciones de hierro bandeado y explorar en más detalle lo que nos dicen sobre la química oceánica y el nivel de oxígeno atmosférico de la Tierra joven. Para ello tomemos un vuelo a Perth, Australia, alquilemos un coche y viajemos 1400 kilómetros al norte hasta el parque nacional de Karijini (antes parque nacional del Hamersley Range). Mientras conducimos nos fijamos en los canguros —saltan como una flecha delante del coche sin hacernos ni caso— y mantenemos un ojo especialmente atento a los trenes de carretera, o camiones de triple remolque. Vemos el polvo que levantan en la lejanía, pero nos echamos a tiempo al arcén y sujetamos firme el volante mientras nos pasan. Circulan deprisa y, por todo lo que puedo decir, no alteran su marcha por nada ni nadie. También mantenemos un ojo en el indicador de la gasolina. Lo que figuran como pueblos en el mapa en esta parte de Australia son en realidad gasolineras. Son pocas y muy separadas. Si te saltas una, puede que no llegues a la siguiente.
El parque nacional de Karijini contiene rocas que se describen geológicamente como pertenecientes a la cuenca de Hamersley. Entramos en el parque y conducimos hasta una de las gargantas que los ríos han cortado a través de los estratos de rocas sedimentarias. Descendemos al fondo de la garganta (lámina 3) y miramos de cerca de la roca. Primero nos impresionan sus colores: rojo sanguíneo alternándose con capas de rojo claro, gris y blanco. Cuando miramos hacia arriba y todo a nuestro alrededor, este capeado es evidente en todas las direcciones. También recordamos que hemos pasado kilómetros y kilómetros de rocas similares según conducíamos hacia la entrada a la garganta.
El color rojo en estas rocas se debe a la alta concentración de minerales de hierro. Estas rocas son ejemplos de las formaciones de hierro bandeado de las que hemos estado hablando, y revelan mucho sobre la química de los océanos de la época en que se formaron. En primer lugar hay que mencionar su amplia distribución en el espacio y en el tiempo.
[87] Dicho en breve, son un tipo común de roca de la Tierra joven. Después está el hierro; estas cantidades masivas de hierro se encuentran típicamente en capas que pueden extenderse a lo largo de grandes distancias de kilómetros o más. En realidad, muchas de las percepciones clave sobre la química del océano y la atmósfera provienen de entender dónde se originó todo este hierro y cómo se abrió camino hasta el suelo marino.
Para comprender esto, necesitamos saber algo de la química del hierro Fe). Hay puro hierro metálico, todos lo sabemos, pero el hierro metálico no suele encontrarse en la superficie de la Tierra, así que no lo consideraremos aquí. Salgamos al patio trasero y echemos un vistazo al cubo de pirita (del capítulo 5) que dejamos a propósito a la intemperie. No quiero revelar demasiado, pero, si ha estado ahí el suficiente tiempo, deberíamos ver minerales de hierro de un marrón rojizo formándose en la superficie. Esto es básicamente óxido. La mayoría del hierro en la superficie de la Tierra se ha convertido —o quiere convertirse— en óxido en la presencia de oxígeno (piensa en un coche tras cinco o diez años de circulación bajo el viento salado de la costa). Se dice que el hierro en esta forma está oxidado, que significa en términos químicos que, comparado con su forma metálica, cada átomo de hierro ha perdido tres electrones. Eso es el ión férrico y se escribe Fe3+. Desde nuestra perspectiva aquí, podemos pensar en ello como: oxígeno + hierro = óxido, y el óxido es inmóvil.
Mis abuelos tenían una casita de campo en la costa del lago Michigan, a la que surtía de agua un pozo profundo. Nos dijeron que no bebiéramos esa agua, lo que para un niño de siete años supone un fuerte incentivo para beberla. En cualquier caso, el agua tenía un peculiar sabor metálico que muchos conoceréis. Yo ponía un vaso de agua recién recogida en la mesa, y en cuestión de minutos un óxido marrón se empezaba a formar en las paredes del vaso. El sabor metálico provenía del hierro en su forma reducida, que significa en términos químicos que cada átomo tiene un electrón más que el hierro en su forma oxidada. Este es el ión ferroso (al que ya encontramos en el capítulo 2) y se escribe Fe2+. El hierro ferroso es soluble y bastante móvil en el agua, pero tal y como mostraba mi experimento infantil, el hierro ferroso reacciona con el oxígeno para formar óxido insoluble.
Esta es la clave para entender las formaciones de hierro bandeado (BIF). Su escala masiva y su naturaleza laminada significan que el hierro fue transportado en forma soluble por las profundidades oceánicas, y por tanto en ausencia de oxígeno. A medida que este hierro ferroso se iba mezclando con las capas superiores del océano, resultaba oxidado, quizá por las mismas bacterias fototrópicas oxidantes de hierro que vimos en el capítulo 2, o quizá por las pequeñas cantidades de oxígeno en las aguas superficiales que producían las cianobacterias (si es que estaban presentes). Para ser franco, no sabemos con certeza cómo era oxidado, pero el resultado neto era una lluvia de óxidos insolubles de hierro cayendo al fondo del mar, y la formación subsiguiente de las BIF. Examinaremos esto en más detalle en el capítulo 9, pero las condiciones anóxicas que se requieren para el transporte del hierro ferroso por el océano profundo requieren a su vez unas concentraciones de oxígeno atmosférico mucho menores que las actuales.
Todas estas pruebas de bajos niveles de oxígeno atmosférico en el eón Arcaico, con ser persuasivas por sí mismas, han cedido el paso en gran medida a otra línea de razonamiento que resultaba tan nueva y tan inesperada que nadie la vio venir. En 1999, mi buen amigo y colega James Farquhar era un
posdoc en el laboratorio de Mark Thiemens en la universidad de California en San Diego. Estaba trabajando a altas horas de la noche midiendo la composición isotópica de los compuestos de azufre en el espectrómetro de Mark. Cuando los resultados salieron en la pantalla, James se aterrorizó. De pronto había algo tan raro que estaba seguro de que había estropeado la máquina. Apagó y se fue a casa con gesto taciturno, pensando en cómo darle la mala noticia a Mark. A la mañana siguiente, con la cabeza más despejada, midió unas cuantas muestras más y encontró que sus observaciones del día anterior eran correctas. Tras medir una serie de controles, se convenció de que la máquina funcionaba bien. Luego pensó un rato en los datos y se dijo que ahora podía decirle a Mark que, más que romper el espectrómetro de masas, había encontrado una forma completamente nueva de entender la dinámica del oxígeno atmosférico en la Tierra primitiva.
[88]Veamos lo que hizo James y lo que percibió. Su objetivo era medir la composición isotópica de los compuestos de azufre en las rocas sedimentarias primitivas. El azufre tiene cuatro isótopos estables:
32S,
33S,
34S y
36S.
[89] En términos de abundancia natural, encontramos la mayoría del azufre en forma de
32S, que supone el 95,02% del total. El
34S es mucho menos abundante, dando cuenta del 4,21% del total; el
33S y el
36S sólo están presentes en proporciones muy pequeñas, del 0,75% y el 0,02%, respectivamente. Durante décadas, los geólogos sólo se habían preocupado de medir la proporción entre
34S y
32S. Esto se debe a dos razones. Una es que esos isótopos son los más abundantes y, por tanto, los más fáciles de medir. La otra es que, según el razonamiento común, no debería importar a qué isótopos miramos (así que por qué no usar los más fáciles). La idea es que, como vimos en el capítulo 6 con el carbono, si algún proceso favorece un isótopo sobre otro (lo que se conoce como fraccionamiento), entonces todos los isótopos del mismo elemento deberían comportarse de una forma predecible; aquí el grado en que cierto isótopo es favorecido (fraccionamiento) depende de la masa del isótopo. Funciona de la manera siguiente. Hay una diferencia de una unidad de masa entre
32S y
33S (33 - 32 = 1), una diferencia de dos unidades de masa entre
32S y
34S (34 - 32 = 2), y una diferencia de cuatro unidades de masa entre
32S y
36S (36 - 32 = 4). En la práctica, si hay algún proceso que fracciona isótopos, el efecto debería ser cerca de la mitad entre
32S y
33S que entre
32S y
34S. Y el efecto entre
32S y
36S debería ser más o menos el doble que entre
32S y
34S. Estas pautas se cumplen en casi todos los procesos biológicos y geológicos que fraccionan isótopos de azufre. Tales fraccionamientos se llaman «dependientes de masa», puesto que la magnitud del fraccionamiento depende de las diferencias de masa entre los isótopos.
Lo que vio James era muy diferente. En las rocas del eón Arcaico (y en cualquiera que tenga más de 2300 o 2400 millones de años), los isótopos no se comportaban de la manera esperada. Los fraccionamientos no eran dependientes de masa. Se desviaban de esa tendencia, generando señales que se describen como independientes de masa. Esta relación se muestra en la figura 7.2. En esta figura se representa un parámetro llamado Δ33S. Este parámetro designa, simplemente, la diferencia entre los fraccionamientos esperados de un comportamiento dependiente de masa y los observados. Si Δ33S es cero, el mundo se comporta como esperábamos, y este es realmente el caso desde 2300 o 2400 millones de años atrás hasta el presente. Esta transición al comportamiento normal será el foco de atención del siguiente capítulo, pero creo que podemos estar de acuerdo en que algo extraño estaba ocurriendo antes de 2300 o 2400 millones de años atrás. Pero ¿qué era?
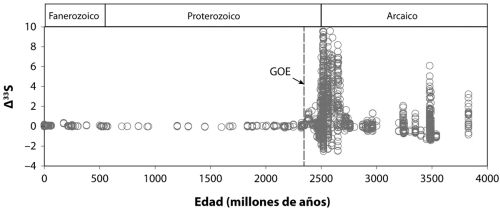
FIGURA 7.2 Compilación de la señal de isótopos de azufre independiente de masa a través de la historia de la Tierra, expresada como Δ33S (detalles en el texto). También se muestran los principales eones de la historia de la Tierra, y el gran suceso de oxidación, o GOE, que será discutido en el próximo capítulo. Compilación de datos cortesía de James Farquhar.
Casi todas las rocas sedimentarias con azufre que tienen más de 2400 millones de años se vieron afectadas por ello, de modo que el proceso (o conjunto de ellos) que produjo esos extraños fraccionamientos fue un fenómeno a gran escala global. Para entender la causa de estos fraccionamientos independientes de masa, tenemos que preguntarnos de dónde vino el azufre y como llegó a los sedimentos. Hoy en día, los ríos suministran la mayoría del azufre a los océanos en forma de sulfato. El sulfato proviene de la erosión de las piritas en presencia de oxígeno y de la disolución de las rocas que contienen sulfato (por lo general yeso), que se formaron en algún momento del pasado por evaporación del agua marina. ¿Recuerdas nuestro viaje en el capítulo 4 con Dave des Marais a la compañía salinera mexicana que contenía aquellas preciosas alfombras de cianobacterias? Durante el viaje recorrimos varios estanques en los que el yeso estaba precipitando, y esto ocurre antes de que el agua marina se haya concentrado hasta el punto de la formación de sal (NaCl).
En cualquier caso, con un buen suministro de los ríos, el sulfato se puede acumular en los océanos hasta unas concentraciones considerables. Quita el oxígeno, sin embargo, y las piritas no se oxidarán a sulfato. Ya vimos evidencias de esto en la cuenca de Witwatersrand y otros depósitos fluviales relacionados del eón Arcaico, donde las piritas permanecieron sin oxidar como arenas de río. Sin una fuente fluvial de sulfato, la concentración de sulfato en el océano se hace muy baja
[90] y, de manera crítica, otras fuentes de sulfato a los océanos se vuelven probablemente significativas.
James siguió esta línea de razonamiento, decidió que una fuente atmosférica de azufre tenía sentido porque el efecto estaría distribuido globalmente y pensó que los volcanes podían constituir una buena fuente de azufre. De hecho, allá por 1962, Dick Holland había reconocido la posible importancia de una fuente volcánica de azufre para el entorno superficial de la Tierra temprana. Si uno mide los gases que emanan de los volcanes actuales, encuentra que el principal gas sulfuroso es el dióxido de azufre, o SO
2. El sulfuro de hidrógeno (H
2S) también está presente en esos gases, y que domine el SO
2 o el H
2S depende —lo que es importante— de la química del manto terrestre en el lugar donde se originan los volcanes. La química del manto, sin embargo, probablemente ha cambiado poco a lo largo de la historia de la Tierra,
[91] de modo que el SO
2 fue seguramente la más importante especie de azufre también en la Tierra primitiva.
Estimulado por su descubrimiento inicial, James condujo una serie de experimentos en los que sometió al SO2 gaseoso a diversas longitudes de onda de luz ultravioleta, que tiene la energía apropiada para transformar el gas SO2 en otros compuestos químicos. James halló que podía generar fraccionamientos independientes de masa de esta forma, y que esos fraccionamientos eran similares a los observados en las rocas sedimentarias del eón Arcaico.
Se puede argumentar, por tanto, que los fraccionamientos independientes de masa observados por James en las rocas arcaicas se produjeron por la interacción de la luz ultravioleta y el gas SO2 surgido de los volcanes. Bien, ahora viene la parte realmente genial. La mayoría de la luz ultravioleta que causa el fraccionamiento independiente de masa del SO2 es absorbida hoy por la capa de ozono de la atmósfera terrestre. El ozono se genera a partir del oxígeno atmosférico. Si quitas el oxígeno, quitas el ozono, y permites los fraccionamientos de azufre independientes de masa observados por James.
Sin embargo, incluso si se produce una señal de isótopos de azufre independiente de masa, es necesario preservarla. Demos entrada de nuevo a Jim Karting, a quien ya encontramos en el capítulo 1, y a su
posdoc de la época Alex Pavlov. Ellos dos abordaron este problema con un complejo modelo de la fotoquímica atmosférica y concluyeron que la preservación del efecto independiente de masa de los isótopos del azufre requiere unos niveles de oxígeno atmosférico 100 000 veces menores que los de hoy (¡eso es <0,001% de los niveles actuales!).
[92] Por tanto, los resultados de James, combinados con el modelado de la atmósfera, nos regalan un importante barómetro del oxígeno antiguo, y ese barómetro nos dice que los niveles de oxígeno en la Tierra joven debieron ser realmente muy, muy bajos. Lo que es totalmente coherente con la visión original de Dick Holland.
Uno podría sentirse tentado de finalizar aquí, pero los científicos, por fortuna, son gente muy curiosa. Es difícil decirles lo que tienen que pensar, y siguen hurgando, sondeando e inquiriendo, tratando de encontrar cosas nuevas. En dos lugares del mundo, dos grupos diferentes estaban examinando a fondo las rocas depositadas en las postrimerías del eón Arcaico. Un grupo, encabezado por Martin Wille, de la Universidad Nacional de Australia, estaba investigando rocas de Sudáfrica con edades entre 2650 y 2500 millones de años. El otro grupo, liderado por Ariel Anbar, de la Universidad Estatal de Arizona, examinaba rocas de 2500 millones de años del oeste de Australia (no muy lejos de las gargantas de Hamersley que visitamos antes). Veamos lo que hicieron.
Estos dos equipos se habían hecho la misma pregunta. ¿Podrían identificar algunas fases minerales sensibles a oxígeno que generaran un producto observable de alguna forma en el registro geológico? Esta estrategia es potencialmente muy sensible. Supongamos, por ejemplo, que tenemos un mineral que se oxida a medias, o incluso sólo un 10%, en un entorno de bajo oxígeno. Aunque oxidado parcialmente, este mineral podría preservarse en un depósito fluvial, como vimos antes, y sería un indicador de bajo oxígeno. Un producto de oxidación, sin embargo, podría también ser liberado, y si fuera descubierto por algún científico curioso demostraría que, aunque a baja concentración, algo de oxígeno estaba presente.
Un buen candidato para explorar es el molibdeno (Mo), y los dos equipos se centraron en él. En la Tierra joven, en ausencia de oxígeno, lo más probable es que el molibdeno estuviera presente en las rocas sobre todo como sulfuro de molibdeno (MoS2) o como un componente menor de la pirita. Estas formas de molibdeno están reducidas químicamente y son estables en ausencia de oxígeno. En presencia de oxígeno, sin embargo, las fases de sulfuro de molibdeno son oxidadas rápidamente a una forma soluble en agua y móvil, el ión molibdato (MoO42-). Este ión, una vez formado, es transportado por los ríos hasta el mar. Por tanto, sin oxígeno en la atmósfera, no hay transferencia de molibdato a los océanos y la presencia de molibdato en el agua marina es despreciable. Añade oxígeno, sin embargo, y la concentración de molibdeno aumentará en el océano.
Ambos equipos buscaron enriquecimientos de molibdeno en los sedimentos primitivos que exploraban, y ambos lo encontraron (figura 7.3). El elemento renio (Re) se comporta de modo similar al molibdeno, y ambos equipos hallaron enriquecimientos de este elemento también. Por tanto, cada equipo descubrió de forma independiente que el oxígeno reaccionaba con compuestos en la superficie de la Tierra en una época en que el registro de isótopos de azufre independientes de masa indica unas concentraciones muy bajas de oxígeno atmosférico.
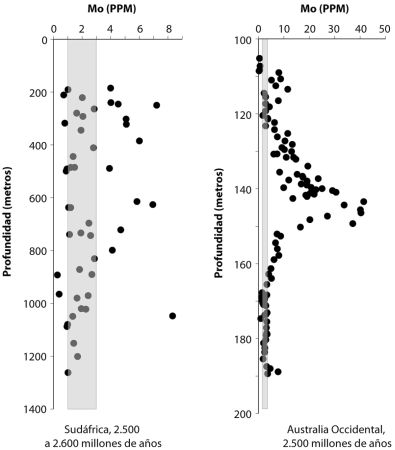
FIGURA 7.3 Concentraciones de molibdeno en las rocas de Sudáfrica y Australia Occidental. Los valores que exceden de la caja gris (promedio para las partículas de la corteza continental y los ríos) representan enriquecimientos y evidencias de «tufillos» de oxígeno durante el eón Arcaico.
¿Significa esto que James estaba equivocado? No. Implica, más bien, que Mo y Re pueden oxidarse y movilizarse en los ríos a concentraciones de oxígeno inferiores a las que se requieren para hacer que desaparezca el efecto isotópico independiente de masa. También implica, y esto es importante, que las cianobacterias andaban por ahí entre 2500 y 2650 millones de años atrás produciendo oxígeno. Si añadimos las pruebas sobre biomarcadores de esteranos presentadas en el capítulo 6, tenemos ahora dos líneas de evidencia independientes de la presencia de cianobacterias en una época en que las concentraciones de oxígeno atmosférico eran todavía muy bajas. Ariel Anbar se refirió a este pulso de oxígeno de las postrimerías del eón Arcaico como un «tufillo» de oxígeno. El nombre ha calado, pero la cuestión es: ¿por qué sólo un tufillo? Si las cianobacterias estaban por allí, ¿por qué las concentraciones de oxígeno atmosférico eran tan bajas?
De nuevo, allá por 1962, Dick Holland dio posiblemente con la idea correcta. En el capítulo 5 introdujimos el concepto de que varios gases reactivos con el oxígeno fueron escupidos por los volcanes y reaccionaron rápidamente con el oxígeno atmosférico. Si la velocidad de introducción de estos gases es lo bastante rápida, arrollarán las tasas de liberación de oxígeno a la atmósfera, que —como vimos en el capítulo 5— vienen controladas por las tasas de enterramiento del carbono orgánico y el azufre de las piritas. Si este es el caso, la liberación de oxígeno a la atmósfera puede ser muy activa, pero el oxígeno no se acumulará a niveles apreciables, pues será destruido químicamente por los gases volcánicos. Como discutimos en el capítulo anterior, no tenemos ni idea de la época en que las cianobacterias evolucionaron por primera vez. Si evolucionaron mucho antes de 2500 o 2600 millones de años atrás, querría decir que la eyección volcánica de gases reactivos con el oxígeno estuvo en claro exceso sobre la tasa de liberación de oxígeno durante mucho tiempo. Y que hace 2500 o 2600 millones de años, sin embargo, la tasa de liberación de oxígeno estaba casi compensada por la tasa de su destrucción por los gases volcánicos. A veces el equilibrio se desplazaba hacia la liberación de oxígeno, generando un tufillo de oxígeno, y a veces el equilibrio se desplazaba hacia su destrucción, y el oxígeno desaparecía.
La cuestión, entonces, se convierte en: ¿cuándo y cómo el oxígeno pasó a ser más que un tufillo y se convirtió en un rasgo permanente de la atmósfera terrestre? Este será el tema del siguiente capítulo.
Capítulo 8
La gran oxidación
Corría el año 1990, y la Universidad de California en Santa Bárbara me invitó a una entrevista de trabajo. Las entrevistas académicas son un asunto extenuante. Suelen llevar dos días enteros de discusiones con los profesores de la facultad, y todo el mundo busca una especie de mezcla sobrehumana de brillantez intelectual y pedagógica. Todo ello combinado con la personalidad y los intereses adecuados para reconciliar a las facciones discrepantes del departamento que llevan años sin hablarse. Como es habitual en estas entrevistas, impartí un seminario del departamento en el que discutí mi trabajo y presenté algunas ideas de por dónde podría ir mi investigación en el futuro. Preston Cloud estaba allí. Por entonces era la sombra de un hombre, debido a su mala salud, pero incluso así su mera presencia me ponía nervioso. Reuní fuerzas y di una charla más o menos pasable (pero no muy buena, no conseguí el trabajo), y Cloud me escuchó con atención todo el rato. Cuando hube acabado, vino hasta mí, me dio la mano y dijo que le había gustado la charla. Intercambiamos unos cuantos cumplidos sobre Yale, donde también él había hecho la tesis, y se fue. No le volví a ver, sólo vivió un año más.
Como era habitual en los científicos de su generación, pero no en los de hoy, el camino de Preston Cloud por la vida académica fue indirecto y variado. Nació en 1912 y, tras acabar el bachillerato en 1929, se alistó en la Armada, donde destacó como boxeador. Se licenció en 1933, durante la Gran Depresión, pero, a pesar de las penalidades económicas de la época, se las apañó para conseguir trabajo por días y pudo pagarse las clases nocturnas en la Universidad George Washington. Pese a tan riguroso horario logró de algún modo acabar en cuatro años, tras lo cual entró en la Universidad de Yale como estudiante de doctorado. Cuando Cloud completó la tesis, impartió clase durante un año en la Escuela de Minas de Missouri, pero fue reclutado en 1941 para el programa de minerales estratégicos coordinado por el Servicio Geológico de Estados Unidos (USGS por sus siglas inglesas). Después de la guerra ocupó un puesto de profesor ayudante en la Universidad de Harvard, pero lo dejó en dos años para volver al USGS como jefe de paleontología; permaneció 10 años en ese puesto. Cloud se trasladó entonces a la Universidad de Minnesota, y fue allí donde surgió su interés por los problemas de la Tierra primitiva. Esto tenía sentido, pues la universidad estaba muy cerca de unas formaciones de hierro bandeado y otras rocas antiguas. En 1965 cambió Minnesota por la Universidad de California en Los Ángeles (UCLA), y tres años después ocupó su último puesto en la Universidad de California en Santa Bárbara (UCSB), donde yo le conocí hacia el final de su vida.
La enrevesada carrera profesional de Preston Cloud, y sus variados puestos, responsabilidades y experiencias, le suministraron sin duda la amplitud y profundidad de entendimiento necesarias para un pensamiento verdaderamente grande. Y pensar en grande fue lo que hizo, a la escala de Vernadsky, en mi opinión. En muchos sentidos, de hecho, Vernadsky y Cloud compartían una visión similar. La interfaz entre la biología y la geología era central para ambos. Sin embargo, la preocupación primaria de Vernadsky era entender de qué forma funcionaba la vida como una fuerza geológica, y su interés por la historia de la Tierra era secundaria. Para Cloud, la historia de la Tierra era lo primario, y estaba especialmente interesado en esclarecer la relación entre la evolución de la vida y la evolución química del entorno de la superficie de la Tierra. En 1968 publicó su primer artículo pensando en grande con el muy académico título Evolución atmosférica e hidrosférica en la Tierra primitiva. Desarrolló sus pensamientos en un artículo de 1972 que lleva uno de los mejores títulos de la historia de las ciencias de la Tierra: Un modelo de trabajo de la Tierra primitiva. Con un título como este, más vale que el artículo sea bueno, y este no fue ninguna decepción.
La tesis de Cloud era que las historias de la evolución química y física de la Tierra estaban entrelazadas. No había mucha evidencia para apoyar esta idea, pero, como gran científico que era, Cloud era capaz de ver patrones y hacer conexiones con la información limitada que tenía. No vamos ahora a viajar por toda la historia de la Tierra, como hizo Cloud. En vez de eso, nos concentraremos en una parte concreta de esta historia.
Empezaremos por donde abandonamos en el capítulo anterior. Es decir, con las evidencias de que las condiciones de bajo oxígeno dominaron todo el eón Arcaico (puntuadas por algunos aparentes «tufillos» de oxígeno hacia el final). Preston Cloud, como Dick Holland antes que él, llegó a esta misma conclusión (sólo que sin los tufillos). Pero Cloud dio un paso más. Si las concentraciones de oxígeno atmosférico eran bajas en el Arcaico, se dijo Cloud, ¿cuándo empezaron a aumentar? Para responder esta pregunta, Cloud recurrió a su experiencia rebuscando entre las rocas justo al norte del lago Hurón en Ontario, mientras trabajaba en la Universidad de Minnesota (figura 8.1). Las rocas aquí forman parte de una gran secuencia de formaciones geológicas conocida como el supergrupo Huroniano, que abarcan en edad de 2500 a 2200 millones de años, y por tanto cubren el periodo de interés. Cloud se dio cuenta de que las más viejas de esas rocas, con edades de 2400 a 2500 millones de años,
[93] contenían antiguos depósitos fluviales con uraninita y pirita residuales, similares a las rocas de Sudáfrica que discutimos en el último capítulo. Sin embargo, subiendo hacia rocas más jóvenes, la uraninita y la pirita desparecían, como lo hacía todo signo de formaciones de hierro bandeado (BIF).
Cloud también percibió una coloración roja particularmente intensa en algunas de las areniscas de la parte alta de la secuencia (figura 8.1), por encima del lugar en que la uraninita y la pirita residuales aparecían por última vez. El color rojo de estas rocas representa hierro en forma oxidada. Sin embargo, estas rocas son muy diferentes de los BIF que vimos antes. En muchos casos, se forman en tierra o en aguas someras y, aunque su coloración roja es prominente, estas capas rojas no están supercargadas con hierro, a diferencia de las que se hallan en los BIF.
[94]Poniendo todos estos indicios juntos, primero Preston Cloud, y después Dick Holland, defendieron que hubo un incremento sustancial en la concentración de oxígeno atmosférico entre 2300 y 2400 millones de años atrás. La uraninita y la pirita desaparecieron al ser completamente oxidadas por los altos niveles de oxígeno atmosférico. Más aún, en opinión de Cloud, los BIF dejaron de depositarse cuando los niveles incrementados de oxígeno penetraron hasta el océano profundo, oxidando el ión ferroso a óxido férrico. Las areniscas rojas, en contraste, se formaron como resultado directo de la erosión en tierra firme bajo una atmósfera oxigenada. Holland bautizó a esa transición a unos altos niveles de oxígeno como «el gran evento de oxidación» (o GOE por sus siglas inglesas). El GOE representa un salto cuántico en el contenido de oxígeno de la atmósfera. Es un asunto de enjundia.
Esta es también una gran historia, pero un problema con la evidencia compilada por Cloud y Holland es que las rocas que contienen uraninita y pirita residuales, como las asociadas a areniscas rojas continentales, no están representadas con continuidad en el registro geológico. Por tanto, nuestra resolución de la cadencia temporal del GOE es bastante pobre a partir de estos indicadores. Hay, sin embargo, otra estrategia que nos permitirá explorar con mucha más precisión el ritmo del GOE. Para seguir esta idea, volvamos a los isótopos de azufre de James Farquhar. Recordemos del último capítulo que la distribución independiente de masa de isótopos de azufre era la norma en el eón Arcaico, y que esos fraccionamientos se habían generado probablemente a concentraciones muy bajas de oxígeno atmosférico. Uno de los mejores aspectos de los isótopos de azufre es que la mayoría de las rocas marinas tienen algún tipo de especie de azufre que podemos analizar. Por lo general es la pirita, pero en algunos casos pueden ser minerales de sulfato. Las rocas marinas están representadas casi continuamente en el periodo que abarca al GOE, de manera que los isótopos de azufre de las especies de azufre asociadas con estas rocas nos deben ofrecer una historia del oxígeno mucho más completa.
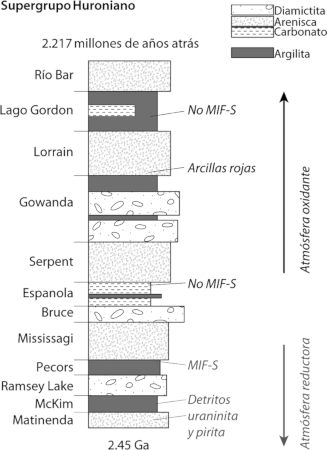
FIGURA 8.1 Estratigrafía general del supergrupo Huroniano. Se muestran los principales acontecimientos de la historia de la oxigenación de la superficie de la Tierra. Nótese que los indicadores del oxígeno indican que el GOE (la gran oxidación) ocurrió después del segundo suceso glacial (representado por la diamictita de Bruce; una diamictita es un depósito glacial). «MIF-S» se refiere a la señal isotópica de azufre independiente de masa, discutida en el capítulo 7. Gráfico tomado de Sekine y colaboradores (2011) con ligeras modificaciones, reproducido con permiso.
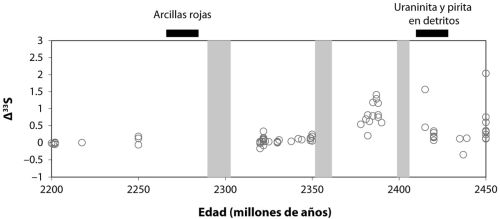
FIGURA 8.2 Cambios en la señal isotópica de azufre independiente de masa (MIF) a través del supergrupo Huroniano. Se muestran también los niveles estratigráficos donde se encuentran las arcillas rojas y las uraninitas y piritas de detritos. Las áreas grises marcan las glaciaciones. Datos isotópicos del azufre cortesía de James Farquhar.
Así que volvamos a las rocas del sur de Notario y midamos los isótopos de azufre. Esto, en realidad, fue lo que hizo Dominic Papineau, del Boston College, y también James Farquhar y su grupo. Además, James y sus colegas analizaron rocas de Sudáfrica de una edad similar, en el llamado supergrupo del Transvaal. Tomados en conjunto, los resultados muestran que la señal isotópica de azufre independiente de masa cambia a la forma normal dependiente de masa entre la última de las uraninitas y la primera arenisca roja (figuras 8.1 y 8.2). ¡Eso es convergencia! Esta transición marca el GOE, al menos en la medida en que afectó a la señal de isótopos de azufre independiente de masa. Nuestra mejor comprensión del fenómeno, por tanto, es que entre 2300 y 2350 millones de años atrás, las concentraciones de oxígeno crecieron hasta un nivel superior al 0,001% del actual, y probablemente, en realidad, hasta mucho más que eso. Ahí lo tienes. Pero no tan rápido: todavía quedan algunas cuestiones importantes. Por ejemplo, aún no hemos explicado qué causó el GOE. Ni hemos discutido cuánto subieron las concentraciones de oxígeno durante el GOE. Más aún, ¿hubo alguna influencia obvia en la biología? Trataremos la primera de estas cuestiones en este capítulo, la segunda en el próximo y la última en el capítulo 10.
Bien, entonces, ¿qué causó el GOE? Para ser sincero, se han propuesto montones de ideas y no las voy a discutir todas. Trataré, sin embargo, de ofrecer una visión de conjunto de las líneas de pensamiento más prometedoras. La hipótesis nula, y la más simple, es que el GOE representa la evolución de las cianobacterias. Tan fácil como eso. Esta idea es promovida con particular energía por Joe Kirschvink, del California Institute of Technology. Joe es bien conocido por romper marcos conceptuales, y ha aportado algunas de las ideas más creativas de las modernas ciencias de la Tierra.
[95] En este debate del GOE, Joe interpreta el papel del abogado del diablo y reconoce abiertamente que sostiene el punto de vista «escéptico». Aborda el problema con la siguiente pregunta: «¿Cuándo demanda la evidencia geológica la presencia de fotosíntesis oxigénica?». Luego considera todas las evidencias que hemos discutido hasta ahora sobre cianobacterias antes del GOE, pero invierte la mayoría de su energía en la evidencia del esterano sobre un entorno con presencia de oxígeno que exploramos en el capítulo 6.
Recordemos que, por todo lo que sabemos, los esteranos tienen un requerimiento absoluto de oxígeno para su síntesis y que, por tanto, encontrarlos es un buen signo de oxígeno en el entorno primitivo. También vimos que la contaminación es una preocupación importante. Joe también levanta esta bandera, pero su principal argumento es que, mientras que la ruta bioquímica conocida de la formación de esterano tiene pasos que requieren oxígeno, este no fue necesariamente el caso durante toda la historia de la Tierra. Joe subraya que los pasos aeróbicos de la síntesis de esterano pueden tener potenciales equivalentes anaeróbicos que no requieren oxígeno. No se sabe de ningún organismo vivo que conduzca esa síntesis de esterano anaeróbico (libre de oxígeno), pero Joe arguye que eso se debe a que las rutas anaeróbicas fueron sustituidas por rutas aeróbicas cuando el oxígeno se hizo disponible. La hipótesis de Joe podría ser correcta, pero es difícil defenderla sin evidencias. Quizá sea una cuestión de estilo, pero yo prefiero alinearme con las explicaciones del registro geológico que se sustentan en rutas y procesos bien documentados. Dicho lo cual, aprecio a aquellos que buscan explicaciones no tradicionales al «conocimiento recibido». Cuando Joe, o quien sea, encuentre una ruta anaeróbica de síntesis del esterano, cambiaré de opinión sin ningún problema.
Luego está el asunto de los «tufillos» de oxígeno que discutimos en el último capítulo. Aquí, en mi opinión, Joe esquiva la evidencia. O explica realmente cómo el molibdeno puede liberarse de los continentes a los océanos en ausencia de oxígeno. Además, ¿cómo se asocian los enriquecimientos de molibdeno con otras evidencias de erosión oxidativa en los continentes como los enriquecimientos en renio? A mí me parece que la mejor forma de explicar la evidencia geológica es aceptar que las cianobacterias evolucionaron antes del GOE, como discutimos en el último capítulo. Por tanto, estamos obligados a buscar otras razones para el incremento de oxígeno en el GOE.
Quizá logremos entender algo más si miramos de nuevo a los controles de oxígeno que exploramos en el capítulo 5. Si recuerdas, el oxígeno es liberado a la atmósfera, en último término, por el enterramiento del azufre pirítico y el carbono orgánico en sedimentos. ¿Tal vez podamos hallar alguna evidencia de esto en el registro geológico? La historia del azufre es interesante, y la recogeremos en el siguiente capítulo, pero de momento acepta mi palabra: no hay ninguna evidencia de que un enterramiento masivo de pirita causara el GOE. ¿Qué tal el carbono entonces? Para examinar esto tenemos que volver a los isótopos. La historia esencial es como sigue. Hay dos formas principales de carbono en la naturaleza. Una es el carbono inorgánico, como el CO
2 en la atmósfera y el ión bicarbonato (HCO
3-) en las aguas naturales;
[96] y la otra es el carbono orgánico, la materia de la vida. El carbono inorgánico entra en el océano fundamentalmente desde los ríos y en forma de bicarbonato, y abandona el océano ya sea como carbono orgánico, los desechos de la vida, ya como algún tipo de mineral de carbonato cálcico, como en las conchas, los corales y la caliza. De forma bien simple, lo que entra en los océanos debe al final abandonarlos. El carbono llega al océano en forma inorgánica y lo abandona ya sea en forma orgánica o inorgánica.
Ahora es cuando llega el asunto de los isótopos. El carbono inorgánico llega al océano con una proporción de átomos C-13 a C-12 que, según asumimos normalmente, no ha cambiado mucho a lo largo del tiempo. Como vimos en el capítulo 6, el carbono orgánico producido por organismos fijadores de carbono como las cianobacterias y las algas (que son los principales productores primarios de materia orgánica en los mares actuales) contiene más C-12 que el carbono inorgánico del que procede. Esto significa que el carbono inorgánico remanente tiene menos C-12 o, en otras palabras, resulta enriquecido en C-13. Cuanto más carbono orgánico retiramos del océano, más enriquecido en C-13 se volverá el carbono inorgánico que queda en el océano. Podemos ver directamente que, si tuviéramos unas tasas muy altas de retirada de materia orgánica del océano, deberíamos generar unos enriquecimientos enormes en C-13 en el carbono inorgánico marino.
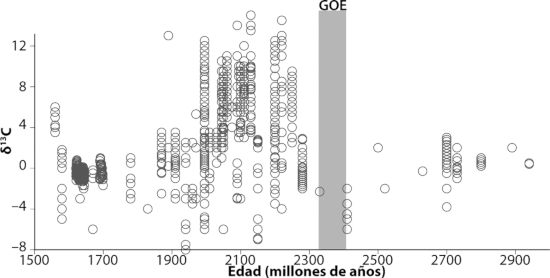
FIGURA 8.3 Composición isotópica del carbono inorgánico, mostrando la anomalía isotópica de Lomagundi que abarca de 1950 a 2300 millones de años atrás. También se muestra la gran oxidación (GOE).
Podemos medir la proporción de átomos C-13 a C-12 de la materia orgánica en rocas viejas, como Minik Rosing hizo en las rocas de Isua, Groenlandia (capítulo 6); y, puesto que el carbono inorgánico también es retirado como calizas (y conchas tras la evolución de los animales), podemos medir la proporción de C-13 a C-12 en el carbono inorgánico también. Así, podemos reunir un registro de la proporción de C-13 a C-12 tanto del carbono orgánico como del inorgánico a lo largo del tiempo. Si recuerdas del capítulo 6, solemos discutir estas proporciones de isótopos de carbono como valores δ13C, y así lo haremos aquí.
Ahora miremos a los datos (figura 8.3) centrándonos en el tiempo del GOE (recuerda que fue de 2300 a 2350 millones de años atrás). De hecho, el δ
13C del carbono inorgánico se vuelve muy elevado alrededor de ese tiempo, y ese periodo de valores altos ha sido bautizado como anomalía isotópica de Lomagundi. Según todas las apariencias, se trata de la mayor anomalía isotópica del carbono de la historia de la Tierra. Esta anomalía fue plenamente apreciada por primera vez en 1996 por Dick Holland y su colega Juha Karhu, de la Universidad de Helsinki.
[97] Estos científicos vieron la anomalía, y su pulso asociado de enterramiento de carbono orgánico, como la fuente de oxígeno que disparó el GOE. Problema resuelto, aparentemente. Sin embargo, si miramos otra vez al gráfico con cuidado, podemos ver que las cosas no acaban de encajar. Una datación reciente y mejorada sitúa la anomalía isotópica de Lomagundi no durante, sino después del GOE. Maldita sea, era demasiado bueno para ser cierto. Estamos obligados a buscar otra causa.
Para presentar la próxima causa posible empecemos con esos fugaces tufillos de oxígeno de las postrimerías del eón Arcaico que discutimos en el último capítulo. Durante esos tufillos, parece como si la atmósfera experimentara pulsos periódicos de oxígeno, sólo para verlos desvanecerse cada vez. También ofrecimos en el último capítulo una explicación tentativa para los tufillos, cuando insinuamos que, en ese periodo de la historia de la Tierra, el flujo de gases reductores desde el manto era similar al flujo de liberación de oxígeno debido al enterramiento del carbono orgánico y la pirita. La mayor parte del tiempo el flujo volcánico estaba en exceso, pero de modo ocasional el equilibrio se desplazaba hacia un exceso de liberación de oxígeno, generando un tufillo de oxígeno en la atmósfera.
Sigamos esta línea lógica, pero, para hacerlo bien, tenemos que empezar mucho más atrás, cerca del principio de la historia de la Tierra. De hecho necesitamos retroceder hasta antes del inicio del registro geológico, hasta una época en la que no tenemos más guía que nuestro ingenio y nuestras mejores suposiciones. Lo que queremos saber es la velocidad a la que los gases reactivos con el oxígeno, sobre todo el hidrógeno (H2), escupido por los volcanes cuando la Tierra era realmente joven.
¿Cómo podemos siquiera aventurar una suposición? Bueno, empecemos por el presente. Tenemos una idea de cuánto gas hidrógeno sale hoy de los volcanes, al menos dentro de un margen de 2 o 3 veces, probablemente. La velocidad de expulsión del hidrógeno dependerá de la química del manto, pero ya vimos en el último capítulo que no es probable que esa velocidad haya cambiado mucho a lo largo de la mayor parte de la historia de la Tierra, así que no nos preocuparemos por eso a partir de ahora. La velocidad de desgaseado (eyección) del hidrógeno debería depender también de la velocidad a la que el material viscoso del manto se mezcla o se mueve por convección. En el capítulo 1 vimos cómo este proceso de convección del manto impulsa la tectónica de placas, que a su vez impulsa el reciclado de materiales en la superficie de la Tierra y permite, básicamente, que la Tierra sea un lugar grato para la vida.
La cuestión entonces se convierte en la siguiente: ¿cómo ha cambiado con el tiempo la velocidad de colección del manto? La tasa de convección es controlada, en una primera aproximación, por el gradiente de temperatura entre el interior de la Tierra y su superficie; cuanto más acusado sea el gradiente, más rápida será la convección. (Puedes ver cómo funciona esto con un simple experimento casero si miras la nota 6.)
[98] El gradiente de temperatura depende a su vez de la tasa de generación de calor en la Tierra. Se estima que el centro de la Tierra está a unos tostadores 5500 ºC (aunque la cifra exacta no se conoce). Parte de esta temperatura tiene relación con el tremendo calor generado cuando la Tierra se condensó por acreción de pequeños
planetesimales hace mucho tiempo.
[99] De hecho, en las fases finales de este proceso, se cree que un objeto masivo, como del tamaño de Marte, golpeó a la Tierra con tal fuerza que le hizo escupir la Luna como resultado. Esta colisión debió de reducir la Tierra a una masa fundida.
El calor también se genera por la desintegración radiactiva de ciertos elementos del manto y el núcleo del planeta. Como los isótopos radiactivos se desintegran hasta formas químicas no radiactivas, la radiactividad de la Tierra ha experimentado una continua disminución con el tiempo, con la consiguiente reducción de la producción de calor.
En suma, la pérdida de calor desde la formación temprana de la Tierra, así como la continua reducción del calor producido por desintegración radiactiva, deben haber resultado en el enfriamiento del interior del planeta con el tiempo. Lo que a su vez habrá ralentizado la tasa de convección del manto. Por tanto, se puede argumentar que la velocidad de liberación de H2 habrá disminuido con el tiempo a causa de una convección cada vez más lenta. La tasa de reducción del flujo de H2 con el tiempo revista una extrema importancia aquí, pero, por desgracia, esta tasa es más bien una conjetura por el momento. Si asumimos, por ejemplo, que la velocidad de liberación del H2 ha decrecido linealmente con el flujo de calor desde el manto, hacemos un conjunto de predicciones, y bastante pobres, por cierto, porque la historia del flujo de calor a través del tiempo no se conoce muy bien. Si la tasa de liberación del H2 ha decrecido en una relación diferente (no lineal) con el flujo de calor, hacemos otro conjunto de predicciones. Con toda franqueza, carecemos del entendimiento suficiente para juzgar qué velocidad de disminución es la más apropiada, pero cierto grado de reducción con el tiempo del flujo de H2 al entorno de superficie es razonablemente seguro.
Pese a estas dificultades, muchos investigadores, incluido yo mismo, hemos tratado de jugar a este deporte. De hecho debemos hacerlo si queremos entender la evolución en el tiempo de la química de la superficie de la Tierra, y la conexión entre el entorno de superficie y el interior de la Tierra es, simplemente, demasiado importante como para ignorarlo. Dick Holland también apreció la importancia de esta conexión e hizo su intento de predecir la historia del flujo de H2 desde el interior de la Tierra a través del tiempo. Esta estimación se muestra en la figura 8.4 con dos líneas que representan el punto de vista de Dick sobre el intervalo de incertidumbre en los cálculos.
Dick también intentó predecir las tasas potenciales de producción de oxígeno por enterramiento de carbono orgánico y pirita a través del tiempo.
[100] No voy a entrar aquí en los detalles de este cálculo,
[101] pero los resultados se presentan también en la figura 8.4. Para interpretar este gráfico hay que centrarse en los dos puntos negros, no en todas las áreas grises solapantes. Estos puntos deben verse como los extremos del cálculo. Lo que vemos es muy iluminador. El modelo de Dick insinúa que en algún momento entre 2000 y 2700 millones de años atrás, la tasa de liberación de oxígeno excedió por primera vez a la demanda de oxígeno. Esta figura ofrece una explicación gráfica de la causa del GOE.
Figura 8.4 Cálculos de Dick Holland sobre el flujo de hidrógeno desde el manto en función de las tasas de liberación de oxígeno a la atmósfera. Los puntos negros indican puntos de cruce en dos escenarios diferentes contemplados por el modelo. Datos tomados de Holland (2009).
¿Es esta realmente la manera en que ocurrió? Pondré la mano en el fuego y diré: «Creo que sí», al menos a grandes rasgos. Simplemente, tiene muchísimo sentido. Siendo justos, hay otros que han tenido ideas similares, en particular Jim Karting (a quien encontramos en el capítulo 1) y Lee Kump, ambos de la Universidad Penn State, y Dave Catling, de la Universidad de Washington. Todos ellos han descrito, como Dick, la forma en que el GOE representa probablemente el punto en el tiempo en que la liberación de oxígeno superó a la demanda de oxígeno, y todos ellos, entre otros, han presentado modelos inteligentes, aunque diferentes, para apoyar esa idea. A mí me gusta en particular el modelo de Dick, sin embargo, debido a su simplicidad y porque los procesos que prescribe tienen un buen sentido geológico.
De modo que Preston Cloud estaba en lo cierto. El registro geológico demuestra que, hace unos 2300 millones de años, el contenido de oxígeno de la atmósfera terrestre se incrementó de manera drástica. Puesto que es probable que las cianobacterias evolucionaran mucho antes, no parece que una atmósfera bien oxigenada fuera una consecuencia necesaria o inmediata de la actividad de los organismos productores de oxígeno. La química atmosférica es esclava de la dinámica del manto, puesto que el interior y el exterior del planeta están conectados de manera profunda. En verdad, llevó la mitad de la historia de la Tierra que el manto se calmara hasta el punto en que el oxígeno pudiera acumularse. Esto, sin embargo, representó un punto de inflexión —o un punto de no retorno, si se quiere— en el que la química de la Tierra se alteró para siempre. En el siguiente capítulo veremos qué vino después.
Capítulo 9
La edad media de la Tierra: después del GOE
¿No es el sueño de cualquiera viajar en una máquina del tiempo? Bueno, quizá no de cualquiera, pero a la mayoría de los geólogos que conozco les encantaría montar en una. Lo que importa aquí para nuestra historia es que, con una máquina del tiempo, podríamos probar directamente si nuestras ideas sobre la historia del oxígeno atmosférico son correctas. Hemos ensamblado esta historia a partir de nuestra lectura del registro geológico, pero ya hemos mencionado que este registro ofrece una visión imperfecta del pasado. Tomamos una roca, un trozo de antiguo sedimento que antaño fue barro en el fondo del mar; tal vez ha sido calentado y eso ha alterado los minerales o la materia orgánica, o quizá los fluidos han penetrado mucho más tarde en la roca, modificando su química. La roca representa la suma de todos los procesos que la han influido durante su larga historia en la Tierra, y no sólo aquellos que nos interesan más.
Además, sólo podemos conjeturar de qué manera las señales en las que estamos interesados se inmovilizaron en la roca. Por ejemplo, podemos comprender estudiando el entorno moderno cómo el molibdeno se abre camino hacia los sedimento (en el capítulo 7 usamos el molibdeno para registrar «tufillos» de oxígeno en la atmósfera). Hacemos esto porque podemos medir la concentración de molibdeno en el agua que cubre los sedimentos de hoy. Podemos trazar su ruta desde ahí hasta los sedimentos, y comprender así qué procesos actúan para controlar esa ruta. Podemos hacer suposiciones y sondear nuestro entendimiento con modelos y experimentos. De hecho, este tipo de trabajo constituye la base de nuestra lectura del registro geológico. Pero, cuando miramos a la roca, lo único que tenemos es la concentración de molibdeno en ella, y tal vez alguna otra especie química, para darnos algunas pistas sobre la química de las aguas primitivas. No tenemos el agua de la que se derivó el molibdeno. Por tanto, a fin de cuentas, sólo podemos estimar su concentración en los antiguos océanos. Esta historia es más o menos la misma para todos los indicadores que usamos para comprender la química del océano primitivo. Nuestra lectura del registro geológico mejora continuamente, pero las pistas son pocas, y nuestras interpretaciones están sujetas a grandes incertidumbres. Realmente, podríamos responder montones de cuestiones con una máquina del tiempo.
El tópico que tratamos ahora son las secuelas del GOE. Pero lo cierto es que, para entender lo que vino después del GOE, tenemos remontarnos con más detalle aún a lo que pasó antes. Ya sé que hablamos del GOE en el último capítulo, y de lo que pasó antes en los capítulos 6 y 7, pero necesitamos buscar cosas pequeñas que se nos pueden haber pasado por alto. Cosas que pueden ser difíciles de determinar a partir de nuestra lectura del registro geológico. Así que usemos nuestra máquina del tiempo y viajemos a la época justo anterior al GOE. Pongámonos las botas de goma (y una máscara de oxígeno, por lo que pudiera pasar) y demos una vuelta por los ríos antiguos. Estamos buscando pirita. Como vimos en el capítulo 7, antes del GOE parte de la pirita se asentaba en forma de guijarros, sin oxidar, en las primitivas cuencas fluviales. Pero muy pocos de esos ríos han sobrevivido, y queremos determinar si el transporte de pirita por los ríos era común o infrecuente. ¿Pudo parte de la pirita haber resultado oxidada? ¿Tal vez sólo los trozos grandes de pirita sobrevivieron a la oxidación, mientras que los más pequeños se perdieron por oxidación? Si pudiéramos retroceder aún más en el tiempo, podríamos seguir la pirita desde su origen en las rocas sometidas a erosión en tierra firme hasta su lugar final de reposo.
Esto nos permitiría decidir cómo la pirita se recicló y se volvió a reciclar. Podríamos ver cuánta de la pirita liberada durante la erosión en tierra se transportó por los ríos, al mar y de vuelta otra vez a unos sedimentos que al final se convertirán de nuevo en piedra. Si la pirita se recicló de esta forma, sin oxidación en una atmósfera de bajo oxígeno, se habría acumulado a concentraciones cada vez más altas en las rocas sedimentarias, a medida que los gases volcánicos continuaban suministrando más y más azufre a la superficie de la Tierra. Ese reciclado también se aplicaría a otros minerales sensibles al oxígeno. Esta secuencia de sucesos tendría mucho sentido en una atmósfera con poco oxígeno, pero ¿fue así como sucedió?
También podríamos examinar el destino de la materia orgánica mientras era erosionada de antiguas pizarras, ya que estas rocas albergan la mayoría de la materia orgánica en el registro geológico. Viajando al pasado, me llevaría un pequeño taladro. Con él recogería muestras que abarcaran desde la superficie de la pizarra expuesta a la lluvia y la intemperie hasta las capas profundas a las que no han llegado aún los fluidos erosivos. Esto me permitiría ver cómo resultó afectada la materia orgánica a medida que los fluidos erosivos penetraban en la roca y la transformaban.
En el mundo moderno, según reveló Steven Petsch y discutimos en el capítulo 5, las concentraciones de materia orgánica son universalmente menores junto a la superficie superior de la pizarra, ya que la pizarra sufre la erosión oxidativa (figura 5.3). Esta oxidación requiere oxígeno, pero no es extremadamente rápida, y queda algo de materia orgánica incluso en la superficie superior que queda expuesta al oxígeno durante más tiempo.
[102] Entonces, ¿qué pasa cuando las concentraciones de oxígeno son realmente bajas, como lo eran antes del GOE? Yo esperaría que la materia orgánica evitara la oxidación mientras las antiguas pizarras se elevaban al entorno erosivo. Si fuera así, la materia orgánica, al igual que la pirita, podría haberse reciclado en buena medida de roca a sedimento y a roca de nuevo, acumulándose en concentraciones cada vez más altas a medida que más y más CO
2 fuera entrando en el entorno de superficie desde los volcanes
[103]. Por tanto, la máquina del tiempo nos habrá dicho algo muy importante sobre la dinámica de las especies minerales sensibles al oxígeno antes del GOE. En ausencia de una máquina del tiempo, sólo podemos hacer suposiciones razonables y esperar que sean correctas. Suponemos, por tanto, que antes del GOE (y dejando aparte los periodos de tufillos de oxígeno que discutimos en el último capítulo), una gran proporción de pirita, materia orgánica y muchas otras especies sensibles al oxígeno traídas al entorno erosivo escapó de la oxidación y fue reciclada y vuelta a reciclar de roca a sedimento y a roca de nuevo.
[104]Ahora llega el oxígeno. Un incremento del nivel de oxígeno atmosférico habría causado una oxidación eficiente de la pirita a ácido sulfúrico,
[105] y de la materia orgánica a CO
2 con la liberación potencial de cualquier nutriente atrapado en la materia orgánica. ¡Esto puede verse como el equivalente geoquímico de agitar un nido de avispas! Dick Holland fue, hasta donde yo sé, el primero que especuló sobre lo que debió de ocurrir después:
La gran anomalía positiva de δ13C en los sedimentos de carbonato entre 2220 y 2060 millones de años atrás indica que, durante este periodo, PO4-3 estaba disponible a niveles excepcionales para la fotosíntesis y el enterramiento de carbono. Es probable que varios cambios contribuyeran a esa disponibilidad incrementada. Cuando el contenido en O2 de la atmósfera se elevó, la oxidación del FeS2 durante la erosión debió incrementarse de manera drástica. El H2SO4 generado durante la oxidación del FeS2 debe haber incrementado la tasa total de erosión química y, por tanto, la tasa a la que el PO4-3 se vertía a los océanos. Al mismo tiempo, el H2S y el HS- en las zonas cercanas a la superficie de los océanos fueron probablemente oxidados a SO4-2. Este proceso redujo el pH de ríos y mares y tal vez hizo al PO4-3 más disponible para la fotosíntesis.
Así pues, Holland arguye que el ácido generado durante la oxidación de la pirita en respuesta al GOE habría reforzado el proceso de erosión. Es bien sabido que la mayoría de las rocas se disuelven más deprisa en condiciones acídicas, pero lo más importante aquí son los minerales concretos de las rocas que contienen fósforo. Un mineral importante de este grupo es la apatita [Ca5(PO4)3(OH,F,Cl)], que es básicamente la misma sustancia que en nuestros dientes y huesos. Como nuestros dientes cuando se exponen en exceso a bebidas carbonatadas, el GOE, según Dick Holland, produjo una deterioro masivo de la apatita en la superficie de la Tierra. Esto, a su vez, liberó enormes cantidades de fósforo a los océanos. Este gran suministro de fósforo estimuló la producción primaria y condujo a la gran tasa de enterramiento de materia orgánica que produjo la masiva anomalía isotópica del carbono de Lomagundi que discutimos en el último capítulo. En una curiosa inversión de causa y efecto, la anomalía de Lomagundi, antaño saludada como la causa del GOE, resulta ser ahora su consecuencia.
Por otra parte, la materia orgánica que se había acumulado en las rocas hasta el GOE estaría acompañada por nutrientes como el nitrógeno y el fósforo. La oxidación de esta materia orgánica tras el GOE habría liberado esos nutrientes también, aportando otra fuente de fósforo a los océanos. Quizá la liberación de todo ese fósforo se benefició también del ácido generado durante la oxidación de la pirita en las rocas ricas en materia orgánica. En suma, me gusta mucho la idea de que la liberación del fósforo acumulado en las rocas ricas en materia orgánica tras el GOE ayudó a disparar la anomalía de Lomagundi. Y me gusta porque no sólo aporta una razón para el inicio de la Lomagundi, sino también para su final.
El lector se podría preguntar, razonablemente, por qué necesitamos una razón para ese final. Se debe al modo de operación del ciclo de las rocas. Ya aprendimos en capítulos anteriores que, por desgracia, el registro geológico de rocas realmente antiguas es escaso. La razón es que, a lo largo del tiempo, han tenido una probabilidad mayor de aflorar a la zona de erosión de la superficie de la Tierra. De hecho, si cuentas todas las rocas que se pueden encontrar en la superficie de la Tierra y las agrupas por edades, hallas que la abundancia de las rocas decrece a medida que su edad aumenta (figura 9.1). Si tratas de cuantificar este efecto, cerca de la mitad de las rocas que quedan de cualquier tiempo en la historia de la Tierra desaparecerán por erosión (aunque algunas están enterradas en la corteza) en una escala temporal de 200 a 300 millones de años. Este viene a ser el intervalo de tiempo de la anomalía isotópica de Lomagundi, y creo que tiene sentido que la duración de ese suceso pueda haber estado limitada por la disponibilidad de viejas rocas del Arcaico y la disponibilidad de materia orgánica reciclada y de nutrientes. Que quede claro que todo esto es especulación, pero apostaría a que el evento de Lomagundi será sometido a un intenso escrutinio en los próximos años, y me sorprendería que el instinto inicial de Holland, que lo relaciona de alguna manera con el GOE, se demostrara incorrecto.
Así, el GOE parece haber sido seguido por un enterramiento masivo de carbono orgánico que se expresa como la anomalía isotópica de Lomagundi. Dick Holland y su antiguo posdoc Andrey Bekker han argumentado que la anomalía de Lomagundi se apoyó en los ya altos niveles de oxígeno del GOE para producir unos niveles de oxígeno similares a los del presente. No hay mucha evidencia de momento para sustentar esto, pero si el oxígeno se volvió así de alto, ¿es que las cosas volvieron a asentarse después del evento de Lomagundi? ¿Qué era, de hecho, lo normal en ese momento de la historia de la Tierra?
FIGURA 9.1 El área de rocas sedimentarias expuestas en función de la edad geológica. Las caídas bruscas en el tiempo indican en parte el enterramiento de rocas en la corteza terrestre; sin embargo, a grandes escalas de tiempo, la pérdida por erosión física y química es probablemente la causa más importante de la caída. Datos cortesía de Bruce Wilkinson (Wilkinson y colaboradores, 2009)
Hace muchos años empecé a reflexionar exactamente sobre ese asunto. La anomalía isotópica de Lomagundi no había entrado todavía realmente en la discusión, pero el GOE sí. El punto de vista dominante, surgido sobre todo de las contribuciones de Dick Holland y Preston Cloud, era que el GOE condujo a unos niveles de oxígeno lo bastante altos como para ventilar con oxígeno el fondo del océano. Así, la idea de que el oxígeno creció a altos niveles durante y después del GOE no es nueva. Esta oxigenación, a su vez, oxidó el hierro de los océanos. De manera que el GOE trajo un final abrupto a las condiciones ricas en hierro de las profundidades oceánicas y por tanto a la deposición de formaciones de hierro bandeado. Los altos niveles de oxígeno post-GOE podrían, por tanto explicar una importante observación geológica: el cese casi completo de deposición formaciones de hierro bandeado que siguió al GOE (figura 9.2). De hecho, esta es la visión que presenté en el último capítulo.
Mientras que esta idea de unos niveles altos de oxígeno post-GOE parecía bastante razonable, había partes de la idea que no me acababan de convencer. Para empezar, yo había utilizado poco antes un modelo simple de química oceánica, introducido por primera vez por Jorge Sarmiento, de la Universidad de Princeton, para explorar cómo unos altos niveles de oxígeno en las aguas profundas del océano podrían responder a cambios en la concentración de oxígeno atmosférico. El modelo es muy simple y constituye una mera aproximación al funcionamiento del océano real, pero el resultado fue muy chocante. Para oxigenar por completo los océanos después del GOE, como Holland y otros habían insinuado, los niveles de oxígeno atmosférico tendrían que haber aumentado hasta el 40% o 50% de los niveles actuales. Quizá Dick hubiera aceptado fácilmente esta conclusión, pero yo no podía. Por ejemplo, y tal como explicaremos en el siguiente capítulo, se estaban acumulando evidencias de un incremento del oxígeno atmosférico en las postrimerías del precámbrico (hace unos 600 millones de años). Esto tendría un difícil encaje si el oxígeno ya hubiera subido en el GOE a unos niveles cercanos a los actuales. Hay también un argumento más débil que veremos en los dos capítulos siguientes.
[106] Este argumento se basa en la idea de que la evolución generalizada de los animales más o menos grandes y móviles, mucho después en la historia geológica, ocurrió mientras el nivel de oxígeno se elevaba lo bastante para satisfacer sus elevadas demandas energéticas.
FIGURA 9.2 Distribución de edad (en gigatoneladas, Gt, siendo una Gt = 109 toneladas) de las formaciones de hierro bandeado (BIF) y las formaciones de hierro granular (GIF). Las GIF son unos depósitos más arenosos y carecen de la estratificación (bandeo) de las BIF. Redibujado a partir de Bekker y colaboradores (2010), y modificado a partir de Raiswell y Canfield (2012).
Esto sería de nuevo incoherente con una precedente subida del oxígeno a los niveles actuales. Por último, tal vez hubiera otra forma de explicar la desaparición de las formaciones de hierro bandeado.
Yo había estado compilando una historia de los isótopos de azufre a través del tiempo cuando trabajaba como investigador en el Instituto Max Panck de Microbiología Marina en Bremen, Alemania. Mi trabajo real era explorar el destino de la materia orgánica en los sedimentos y los diversos procesos microbianos responsables de metabolizarla, pero la historia de los isótopos de azufre resultaba cautivadora, y se podría decir que era mi trabajo nocturno. Lo que emergió de esta compilación fue un cuadro de la evolución del ciclo del azufre a lo largo de la historia de la Tierra, y un rasgo llamativo era el gran incremento en la diferencia isotópica entre el sulfato y el sulfuro alrededor del GOE (recuerda los isótopos del azufre del capítulo 7). La verdad es que Eion Cameron, del Servicio Geológico de Canadá, fue el primero en reconocer este incremento, pero la nueva compilación subrayaba su singularidad (figura 9.3). Eion había insinuado que el salto se debía a un incremento en la concentración de sulfato del océano en respuesta al GOE y a la oxidación reforzada de los minerales de sulfuro a sulfato durante la erosión en tierra firme. Esto es muy coherente con lo que discutimos en el último capítulo. La lógica aquí es que las bacterias reductoras de sulfato reducen preferentemente el isótopo ligero del azufre en el sulfato, 32S, en comparación con el isótopo pesado, 34S, como vimos en el capítulo 7, pero el grado al que lo hacen es mucho menor cuando la concentración de sulfato es muy baja.
[107] Con mi compilación en la mano, por tanto, yo no podía por menos que coincidir con Eion, pero ahora podía llevar el argumento un paso más allá. Un flujo incrementado de sulfato a los océanos debería estimular la reducción de sulfato y producir más sulfuro en el océano. Como resultado, más hierro disuelto sería retirado para la formación de pirita (FeS
2).
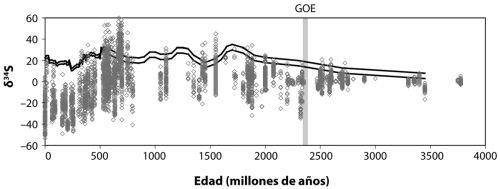
FIGURA 9.3 Compilación de los isótopos de azufre a través del tiempo. Los rombos representan análisis individuales de los sulfuros sedimentarios, mientras que las líneas paralelas reflejan estimaciones de la composición isotópica del sulfato en el agua marina. También se muestra la gran oxidación (GOE). Modificado a partir de Raiswell y Canfield (2012).
Así que, poniendo todo junto, razoné que el GOE probablemente incrementó el flujo de sulfato a los océanos mediante la erosión oxidativa de los sulfuros en tierra firme, reforzando por tanto las tasas de reducción de sulfato a sulfuro de hidrógeno en el océano. El GOE, sin embargo, al menos desde mi punto de vista, no produjo el suficiente oxígeno para oxigenar los océanos. Por tanto, el hierro disuelto no fue retirado de los océanos por reacción con el oxígeno, como supusieron Holland y Cloud, sino por reacción con el sulfuro. Así, según mi propuesta, las profundidades del océano permanecieron anóxicas y se volvieron ricas en sulfuro, en lugar de volverse bien oxigenadas. Andy Knoll, de la Universidad de Harvard, enseguida bautizó a ese estado oceanográfico rico en sulfuro como «océano de Canfield» y, para bien o para mal, el nombre ha calado. En el momento en que hice esa propuesta, sin embargo, no había ni una brizna de evidencia para respaldarla, de modo que devino una alta prioridad buscar evidencias para apoyar o refutar el modelo del océano de Canfield y, de forma más general, definir el estado de la superficie de la Tierra y de la química oceánica después del GOE.
Fue más o menos entonces cuando Simon Poulton vino como
posdoc a mi laboratorio. Simon había trabajado como estudiante de doctorado con mi viejo amigo Rob Raiswell, de la Universidad de Leeds
[108] y, al igual que Rob, Simon sabe orientarse en un
pub. También como Rob, es un científico creativo e ingenioso. De manera que, para explorar el problema del océano de Canfield, Simon y yo nos centramos en el último episodio de deposición de formaciones de hierro bandeado antes de su resurgimiento mucho más tarde en la era Neoproterozoica, que discutiremos en el próximo capítulo. Nuestro foco de atención, por tanto, devino un episodio principal de deposición de formaciones de hierro bandeado ocurrido entre 2100 y 1900 millones de años atrás, que puede observarse en varios lugares alrededor del mundo, pero con una representación especialmente buena en el norte de Minnesota y el sur de Ontario. Ese se convirtió en nuestro objetivo.
Pero espera un momento, dirá el lector, con independencia del modelo, yo pensaba que el GOE era lo que había traído el final de las formaciones de hierro bandeado, ¿y ahora me vienes con unas formaciones que ocurrieron 300 o 400 millones de años después del GOE? ¿De qué vas? Es una buena pregunta y, antes de que podamos entrar en nuestro trabajo sobre el modelo del océano de Canfield, tenemos que lidiar con estas formaciones de hierro bandeado.
Lo cierto es que estas formaciones son muy reveladoras. Para empezar, su deposición empezó justo después o cerca del final de la anomalía isotópica de Lomagundi. Además, al menos parte de ellas se depositaron en unas aguas muy poco profundas. ¡Imagina que te das un baño en la playa y vuelves a casa cubierto de óxido! Este punto es crítico. Puedes imaginar así el escenario: el hierro disuelto, acumulado en el océano profundo libre de oxígeno, fue transportado hacia arriba hasta la plataforma continental mediante un proceso llamado surgencia (
upwelling), y luego se desplazó por la plataforma hasta las aguas someras.
[109] Esto suena bastante simple, pero, si recordamos el capítulo 7, el Fe2+ reacciona deprisa con el oxígeno para formar óxido. Por tanto, si las concentraciones de oxígeno fueran altas como hoy, el Fe2+ simplemente no podría ser transportado desde el océano profundo, a través de la plataforma continental hasta la playa. La única forma lógica que se me ocurre de que el Fe2+ persista a través de un largo transporte por la superficie del océano es que el nivel de oxígeno sea muy bajo, quizá sólo del 0,1% de los niveles actuales o incluso más bajo.
[110] De hecho, hay datos geoquímicos recientes que aportan evidencias independientes de un bajo nivel de oxígeno en esa época. Estas evidencias se refieren al comportamiento de los isótopos de cromo. No entraré en ellas aquí, pero puedes aprender más sobre ello siguiendo la nota 10.
[111]Repasemos. Hubo un incremento sustancial de oxígeno que definió el GOE. Esto puede haber conducido a su vez a la anomalía isotópica de Lomagundi que, como argumentamos antes, estuvo asociada con unas altas tasas de enterramiento de materia orgánica y, tal vez, unas concentraciones de oxígeno aún más altas. Esta anomalía fue seguida poco después por el desplome del oxígeno hasta unos niveles muy bajos y un regreso a la deposición de formaciones de hierro bandeado. Se puede conjeturar que esos bajos niveles de oxígeno fueron, de hecho, una consecuencia de la anomalía de Lomagundi. Es fácil imaginar que, una vez que las cantidades masivas de carbono orgánico enterrado durante la anomalía llegaron al entorno erosivo,
[112] habrían representado un gran sumidero de oxígeno, rebajando los niveles de oxígeno atmosférico. Parece entonces que las concentraciones de oxígeno dibujan unos verdaderos dientes de sierra, disparados aparentemente por el GOE.
Ahora volvamos con Simon. Si recuerdas, estábamos interesados en cualquier posible evidencia que apoye o refute la idea de que, con un incremento de los niveles de oxígeno atmosférico, el sulfuro se extendió por las aguas marinas y puso final a las formaciones de hierro bandeado. Como ya hemos mencionado, las rocas del norte de Minnesota y el sur de Ontario se volvieron nuestro foco de atención y, en particular, nos centramos en la formación de hierro de Gunflint, depositada hace unos 1900 millones de años. Como acabamos de argumentar, esta formación —y otras de la misma época— parecen haberse depositado durante un periodo de muy bajas concentraciones de oxígeno atmosférico. Pero ¿qué pasó justo después de la Gunflint parara de depositarse? Para abordar este problema, contactamos con el efervescente Phil Fralick, de la Universidad de Lakehead en Thunder Bay, Ontario. Es probable que Phil conozca la formación de hierro de Gunflint mejor que nadie. Durante un viaje memorable, Phil nos llevó por la región para explorar tanto la Gunflint como los sedimentos que se depositaron justo después. Lo que más nos llamó la atención en el campo fue que la Gunflint daba lugar a unas pizarras muy negras conocidas como la formación Rove. Esto resultaba estimulante, ya que las pizarras negras se depositaron a menudo en aguas sulfurosas.
[113]Las rocas que vimos en el campo eran geniales para entender la relación entre la Gunflint y la Rove, pero no tan buenas para hacer química. Como hemos visto, estas rocas yacen expuestas a la intemperie y la lluvia, oxidándose en el proceso. Cualquier pirita que hubiera estado inicialmente en las rocas, por ejemplo, podría muy bien haberse perdido por oxidación. Por fortuna, Phil conocía rocas frescas, extraídas recientemente del subsuelo, que cubrían maravillosamente la transición de la formación de hierro de Gunflint a la posterior formación Rove. La muestra de roca, por cierto, provenía de un curioso granjero que dispone de un aparejo de perforación y que, un par de veces al año, extrae una muestra de las rocas que subyacen a sus campos para buscar metales preciosos. Cuando las muestras no contienen nada de interés, las dona a la consejería de Desarrollo y Minas del Norte de Ontario. La decepción del granjero era nuestra fortuna y, tras tomar unas muestras de sus extracciones, volvimos al laboratorio para someterlas a una batería de análisis químicos.
No me extenderé en los detalles de los análisis, pero sometimos las muestras a diversos tratamientos químicos.
[114] Nuestros análisis, en definitiva, nos dijeron que las aguas enriquecidas en Fe2+ donde precipitó la formación Gunflint dieron paso a unas aguas sulfurosas de las que se depositó la formación Rove. Esto era muy coherente con el modelo del océano de Canfield. De hecho, más o menos al mismo tiempo que presentamos esto, Gail Arnold estaba trabajando con Ariel Anbar, de la Universidad Estatal de Arizona, y Tim Lyons, de la Universidad de California Riverside (y un antiguo colega de doctorado en Yale) y presentó apoyos adicionales para el modelo del océano de Canfield basados en los isótopos del molibdeno preservados en pizarras negras de 1600 millones de años. Los interesados en cómo funciona este sistema de isótopos pueden seguir la nota 14.
[115]La historia parecía estar tomando forma, pero uno nunca debe sentirse demasiado cómodo con una idea. En un golpe de suerte, Phil Fralik identificó una serie de muestras que representaban entornos que cubrían desde las aguas someras donde se depositaron las formaciones Gunflint-Rove hasta muy lejos mar adentro, donde las aguas alcanzaban más de 100 metros de profundidad. Había un total de 100 kilómetros entre las muestras. Tras un trabajo concienzudo, Phil pudo trazar líneas de tiempo entre todas esas muestras, y Simon pasó casi un año esclareciendo todos los datos geoquímicas, pero al final mereció la pena. Lo que emergió fue un cuadro espectacular y único de la química oceánica que se extendía desde la costa hasta muy mar adentro (figura 9.4). Como vimos antes, la formación Gunflint dio paso a la sulfurosa Rove junto a la costa. Al movernos mar adentro, sin embargo, vimos que la anoxia persistía, pero también vimos que las aguas sulfurosas desaparecían. De hecho, las aguas sulfurosas daban paso a aguas que contenían Fe2+, algo similar (aunque no del todo, como veremos más abajo) a las aguas de las que se depositó la Gunflint. Esas aguas enriquecidas en Fe2+ se llaman «ferruginosas». Esta es una impresionante representación bidimensional a la química oceánica del pasado profundo. ¡Quitémonos el sombrero ante Simon y Phil!
Otros científicos han contribuido también a esta historia, incluidos el grupo de Andy Knoll, Andrey Bekker y sus colegas, así como Tim Lyons y su equipo. En conjunto podemos pintar un cuadro que dice sí, había unas condiciones marinas más sulfurosas que antes del GOE, pero con un montón de evidencias, también, de condiciones ferruginosas en aguas profundas. Y las condiciones ferruginosas se pueden encontrar en todas las épocas posteriores hasta el tiempo en que evolucionaron los animales, 1300 millones de años después. Veremos esa aparición de los animales en el próximo capítulo, pero reflexionemos ahora sobre nuestra posición respecto al modelo original del océano de Canfield. Después del GOE, el océano profundo parece haber permanecido mayormente anóxico, como predecía el modelo, y las condiciones sulfurosas se extendieron, también como el modelo insinuaba. Sin embargo, esas condiciones sulfurosas no eran particularmente amplias. Eran más bien las condiciones ferruginosas las que parecían dominar en el océano anóxico.
Si las condiciones ferruginosas dominaron, ¿por qué son las formaciones de hierro bandeado tan extremadamente raras durante este periodo de la historia de la Tierra, como vimos gráficamente en la figura 9.2? Buena pregunta. Creo que el modelo original del océano de Canfield sigue ofreciendo la respuesta. Tal y como indicaba el modelo, la oxigenación incrementada condujo a más erosión de la pirita y a un flujo mayor de sulfato hacia los océanos. Esto condujo a más producción de sulfuro por reducción del sulfato, extendió las condiciones sulfurosas y reforzó la retirada del Fe2+ de la solución. Incluso así, en general el flujo de Fe2+ al océano era aparentemente mayor que las tasas de producción de sulfuro por reducción del sulfato.
[116] De este modo, mientras que el sulfuro se podía acumular en aquellas regiones del océano en que las tasas de reducción del sulfato eran altas, en otras regiones el Fe2+ dominaba, generando aguas ferruginosas. Parece, sin embargo, que había bastante sulfuro producido por reducción de sulfato para reaccionar con una cuantiosa proporción del Fe2+ disuelto en los océanos. Esto significaba que, mientras que las condiciones ferruginosas podían aún persistir en grandes volúmenes del océano anóxico, las concentraciones de Fe2+ eran significativamente menores que en los tiempos en que las formaciones de hierro bandeado se generaban activamente; simplemente, con menores concentraciones de Fe2+, no había el suficiente hierro disuelto en el océano para sustentar una activa deposición de esas formaciones. Parece, por tanto, que el modelo del océano de Canfield era correcto en general, aunque no en todos los particulares.
FIGURA 9.4 Desarrollo de la química oceánica a través de la transición desde la formación de hierro de Gunflint (junto a la costa de Biwabik) hasta la superpuesta formación Rove (la formación Virginia). En el gráfico, el tiempo transcurre hacia arriba (los sedimentos más jóvenes están arriba), y nos alejamos de la costa a alta mar de izquierda a derecha. Reproducido de Poulton y colaboradores (2010).
Quizá de forma más crucial para la presente discusión, sin embargo, es que unas aguas profundas anóxicas parecen haber sido el estado normal durante la edad media de la Tierra. Esto implica que las concentraciones de oxígeno atmosférico eran considerablemente más bajas que hoy. Pero ¿cuánto más bajas? El modelado original del océano que me introdujo en este campo insinuaba que vastas extensiones de profundas aguas oceánicas anóxicas podrían desarrollarse si el oxígeno atmosférico cayera al 40% o 50% de los niveles presentes. Por desgracia, el registro geológico no aporta una estimación más precisa. Yo creo, sin embargo, que los niveles de oxígeno eran mucho más bajos que eso, probablemente de menos del 10% del nivel presente. Esta aserción no se basa tanto en las evidencias que hemos discutido hasta ahora como en lo que viene en los siguientes dos capítulos.
Capítulo 10
El oxígeno proterozoico y el auge de los animales
La península de Avalon en el sureste de Terranova es un extraordinario lugar. Tallada en hielo glacial, su terreno escabroso fue después esculpido por el viento, la lluvia y el mar. En la península de Avalon nunca te sientes lejos del mar. También es un lugar de tradición, de tradiciones viejas. Ya en 1534, el célebre navegante francés Jacques Cartier afirmó que los bancos de peces eran tan espesos en la costa de Terranova que «frenaban nuestros barcos en el agua». No fue el primero, sin embargo, que se fijó en la abundancia de peces allí, ya que los asentamientos de pescadores se habían establecido en la península de Avalon a principios del siglo XVI. Siguió en 1610 una colonia en la bahía de la Concepción (Cala Cupar) en el norte de la península, y en 1621 se formó otra colonia en Ferryland, al sur de la península (figura 10.1). La última, que se halla ahora en plena excavación geológica, fue establecida por sir George Calvet, después lord Baltimore, y se convirtió en el primer asentamiento colonial en Terranova. En realidad, sir George se mudó al asentamiento en 1627 y lo abandonó el verano siguiente, explicando al rey Carlos I:
He aprendido por costosa experiencia, que otros hombres movidos por privados intereses me habían encubierto, que desde mitad de octubre hasta mediado mayo muestra el invierno su cara más triste a este país, con el mar y la tierra congelados ambos las más de las veces hasta el punto de volverse impenetrables, sin planta ni cosa vegetal alguna que asome de la tierra hasta principios de mayo, ni un pez en el mar, por no hablar de ese aire de una frialdad tan intolerable que apenas cabe soportarlo.[117]
Mucho después, de la mitad del siglo XVIII a la del XIX, los irlandeses, tal vez más adaptados a las circunstancias difíciles, emigraron en masa a Terranova, y sobre todo a la península de Avalon. En 1836, una revisión del Gobierno reveló que la mitad de la población de Terranova era de ascendencia irlandesa, con la mayoría de ellos viviendo alrededor de St John’s. Todavía hoy se puede ver la influencia irlandesa, sobre todo en la parte sureste de la península, la llamada Costa Irlandesa. Aquí gusta la música irlandesa, y los residentes hablan con un acento irlandés de tal carácter que, en muchos casos, aún puede trazarse hasta el condado irlandés original en que los ancestros vivieron hace casi 200 años.
FIGURA 10.1. Mapa de la península de Avalon, en Terranova. Se destacan los lugares discutidos en el texto. La línea gruesa gris marca la llamada «costa irlandesa».
Es aquí, en el pueblecito de Trepassey del sur de la península, donde Simon Poulton y yo hacemos entrar en calor a nuestros cuerpos helados con unas tazas de café bien caliente y una deliciosa sopa de pollo preparada por Paula Carew, una mujer bajita y burbujeante, y propietaria del First Venture, toda una tradición de la gastronomía de Trepassey. Hoy hemos estado en el campo demasiado tiempo, y hasta demasiado tarde. Paula se deja flotar hasta nuestra mesa y nos pregunta con un intenso acento irlandés si querríamos tomar tras el café algo más fuerte, una cerveza o tal vez un whisky. Asentimos con la cabeza de manera enfática a ambas cosas. Formamos parte de un viaje de campo organizado por Guy Narbonne, del Queens College de Ontario, y Jim Gehling, del Museo del Sur de Australia en Adelaida. Guy y Jim son expertos mundiales en un grupo fascinante de organismos marinos que aparecen en el registro geológico hace unos 580 millones de años. Elkanah Billings, un eminente paleontólogo canadiense, fue el primero en describir un miembro de este grupo en 1871. De hecho, encontró un tipo de fósil llamado Aspidella terranovica a simple vista en pizarras negras que afloraban en plena calle Duckworth, en el centro de St John’s. Todavía hoy las puedes ver allí. Billings también se dio cuenta de que esos fósiles pertenecían a estratos sedimentarios depositados antes de la aparición de abundantes animales en el periodo cámbrico, un tópico al que volveremos más adelante.

Figura 10.2 Varios fósiles de la fauna de Ediacara de Mistaken Point, Terranova. A) Charniodiscus, B) rangeomorfo en forma de huso, C) otro tipo de rangeomorfo en forma de huso. Fotos del autor.
Los fósiles de este periodo ganaron prominencia mucho después, sin embargo, cuando Reg Sprigg se topó en 1946 con unas extrañas formas fósiles que interpretó como primitivos animales parecidos a medusas, en las colinas de Ediacara de las montañas Flinders, en el sur de Australia. Estos fósiles de las postrimerías del precámbrico, y muchos otros de una edad similar, han pasado a conocerse colectivamente como la fauna de Ediacara. Tomada en conjunto, esta fauna representa un amplio rango de muy diferentes tipos de organismos, pero que también muestran atributos comunes (figura 10.2). Con un intervalo de tamaños que abarca de un centímetro a varios decímetros, vivían en el fondo marino ya fuera tumbados o apoyados en algún tipo de tallo o pedúnculo, eran esencialmente inmóviles, exhibían morfologías complejas pero semirregulares y, con algunas excepciones, eluden una clasificación obvia en cualquier grupo de animales, modernos o antiguos. Por tanto, la primera impresión de Sprigg de que se trataba de medusas primitivas era probablemente errónea, lo que deja abierta la cuestión de qué representa realmente la fauna de Ediacara.
No hace falta decir que el debate científico es muy activo en este asunto. A pesar de sus formas singulares, hasta los años ochenta se pensaba que la fauna de Ediacara representaba unos tipos de animales antiguos, primitivos. El debate se encendió cuando el gran paleontólogo Dolf Seilacher, de Tubinga, Alemania, reinterpretó los fósiles como algo totalmente diferente. Argumentó que, en lugar de animales, eran variedades extintas de organismos vivos, testigos fósiles de unos linajes fallidos que no dejaron sucesores. En opinión de Dolf, no tenían nada que ver con los animales en absoluto. Dolf tiene muy buen ojo para deducir cómo los animales extintos hace mucho excavaban, se arrastraban y se alimentaban en los fangos del mundo antiguo, y lo que dijo Dolf, por tanto, se tomó muy en serio. Sí, en serio, aunque no todos, quizá ni siquiera una mayoría, están convencidos de su reinterpretación.
De hecho, una corriente de pensamiento reciente ha vuelto a interpretar como animales la mayor parte de estos organismos, si bien no necesariamente pertenecientes a grupos animales con representantes actuales. Por ejemplo, tomemos uno bien raro: un miembro de la fauna de Ediacara llamado
Charniodiscus, que se encuentra en la península de Avalon entre las mismísimas rocas que Simon y yo habíamos ido a explorar (figura 10.2). En vida medía 10 centímetros o más de longitud, y estaba firmemente arraigado al fondo marino. Estaba constituido por un rizoide (sujeción) y un tallo conectado a una fronda que ondeaba suavemente en las corrientes oceánicas, posiblemente capturando pequeñas partículas o materia orgánica disuelta en el agua. Vivían a bastante profundidad, y por tanto fuera del alcance de cualquier rayo de sol. En resumen, estos organismos eran grandes, exhibían una morfología compleja construida con muchos tipos celulares diferentes, como los animales, y comían como los animales. Según Andy Knoll, es difícil imaginar cómo un organismo semejante pudo haber crecido sin una cubierta de células epiteliales,
[118] otro atributo de los animales. Por último, debido a las oscuras profundidades del agua en que vivían, cualquier afiliación con las plantas o las algas queda excluida. Así que ahí lo tienes: muchos atributos que parecen animales, pero no muy parecido a ningún animal de los que conocemos. Y lo mismo ocurre con gran parte de la fauna de Ediacara.
[119]Bien, ¿qué nos había traído a Simon y a mi a la península de Avalon? Resulta que las rocas de la península de Avalon albergan a los representantes más antiguos conocidos de la fauna de Ediacara. Se trata de los llamados rangeomorfos (figura 10.2), que llegan a datar de 575 millones de años atrás y aparecieron relativamente pronto tras el fin de la glaciación Gaskiers, que ocurrió hace 580 millones de años.
[120] ¿Fue esto una coincidencia? ¿Podríamos identificar algún detonante geoquímico que correlacione con —o incluso ayude a explicar— la repentina aparición de formas similares a animales? Nuestro foco de atención estaba en el oxígeno. Ha habido un prejuicio persistente, desencadenado por un artículo publicado en 1959 por Ralph Nursall, de la Universidad de Alberta, que sostiene que la evolución de los animales, o al menos de sus variedades grandes con una demanda de oxígeno relativamente alta, fue espoleada por un incremento en el contenido de oxígeno de la atmósfera. Se puede discutir largo y tendido sobre la relación entre la evolución animal y el oxígeno, y mantendremos esa discusión más abajo, pero Simon y yo razonamos que buscar signos de cambios en la química oceánica sería un buen punto de partida. Esas rocas de la península de Avalon parecían un buen sitio para empezar.
Resulta extraordinario que, a lo largo de la parte sur de la península de Avalon, se puedan tomar muestras de rocas de manera más o menos continua desde un tiempo anterior a la glaciación Glaskiers, a través de la Glaskiers y hasta 20 millones de años después de su final. De manera importante, es posible muestrear con una referencia constante a la emergencia de la fauna de Ediacara, ya que los fósiles son abundantes y están bien preservados a lo largo de estas rocas. Más aún, Guy y Jim son unos directores de viaje de ensueño. Han trabajado en estos primitivos sedimentos durante años, y los conocen probablemente mejor que nadie. Conocen los fósiles, dónde encontrarlos y en qué lugar del fondo marino vivieron esos organismos. Mantuvieron muchas discusiones animadas sobre la forma de comer de estos organismos, sus posibles afinidades con los animales y la manera en que quedaron preservados. Llegamos al famoso enclave Mistaken Point (punto equivocado) cuando el sol ya se ponía. Este es sitio de fósiles mejor conocido de la península de Avalon. A Guy le gusta llegar aquí al atardecer porque en muchos casos estos fósiles, y los del resto de la península, sólo son débiles impresiones en la roca. Difíciles de ver a mediodía, emergen en toda su gloria al proyectar pequeñas sombras bajo la tenue luz del sol poniente.
Simon y yo recogimos cientos de muestras.
[121] De vuelta al laboratorio, hicimos el mismo tipo de análisis químicos, la llamada
especiación Fe, que describí en el último capítulo para la transición entre la formación de hierro bandeado Gunflint y la superpuesta formación Rove. Encontramos que, antes de y durante la glaciación Glaskiers, las aguas profundas del océano en esta parte del mundo eran anóxicas y ferruginosas.
[122] De hecho, esto se puede ver sin más que mirar a los depósitos glaciales de los Gaskiers, que son de un rojo sangre debido al hierro (lámina 9). Trabajos adicionales de Simon, yo mismo y muchos otros han mostrado que esas condiciones ferruginosas del agua profunda eran comunes, y que llegaban a datar de 850 millones de años atrás, como mínimo.
[123] En verdad, esta agua ferruginosas pueden enlazar con la ocurrencia común de condiciones anóxicas y a menudo ferruginosas, como las descritas en el último capítulo, durante la parte media del eón proterozoico. Sin embargo, y de manera crítica, Simon y yo, junto a Guy, encontramos que, justo después de la glaciación Gaskiers, las aguas oceánicas profundas de la península de Avalon se volvieron bien oxigenadas. Esta es, que yo sepa, la primera evidencia de oxigenación del océano profundo durante la larga historia de la Tierra.
[124] Y puede no haber sido sólo un suceso local. En rocas del oeste de Canadá se han hallado evidencias adicionales de la oxigenación de las aguas profundas en la misma época, en trabajos de Yanan Shen, en colaboración con Andy Knoll y Paul Hoffman de Harvard, y también de nuestro grupo.
Por tanto, la fauna de Ediacara de la península de Avalon surgió en un océano que estaba oxigenándose. De hecho, otras líneas de evidencia parecen apoyar esa conclusión. Por ejemplo, el grupo de Tim Lyon ha encontrado muy recientemente que la concentración de molibdeno en las pizarras negras creció drásticamente hace 630 millones de años, en las postrimerías del neoproterozoico, lo que predata de alguna manera nuestras evidencias de oxigenación de las profundidades del océano (figura 10.3). Este incremento en la abundancia de molibdeno implica una elevada concentración de molibdeno en el agua marina, que significa a su vez una retirada menos eficiente del molibdeno del agua marina. La ruta más eficaz de retirada del molibdeno ocurre en condiciones anóxicas, sobre todo durante la interacción con el sulfuro disuelto. Poniendo junto lo anterior, un incremento de la abundancia de molibdeno en las pizarras negras implica menos retirada anóxica del molibdeno de los océanos. Lo que puede a su vez asociarse razonablemente con un incremento de la oxigenación del océano. Lo mismo se concluye de los isótopos del molibdeno (véase la nota 14 del capítulo 9), que muestran un gran salto en la misma época, como detectó mi posdoc Tais Dahl y la entonces estudiante de doctorado Emma Hammarlund (figura 10.3). Tais y Emma también interpretan este salto como un efecto de la retirada alterada de molibdeno en condiciones óxicas, y por tanto un aumento de la oxigenación del océano.
Distintos indicadores geoquímicos aportan unas respuestas algo diferentes, pero un incremento en la oxigenación oceánica parece evidente en la era neoproterozoica tardía, empezando quizá tan pronto como 630 millones de años atrás, y expresándose en indicadores locales de oxigenación 580 millones de años atrás. Todo esto es más o menos coetáneo con la emergencia de los animales macroscópicos. Una posible forma de explicar la emergencia de los animales grandes sería por un incremento de las concentraciones de oxígeno atmosférico. Seguiremos entonces esta línea de razonamiento e intentaremos bregar con tres asuntos relacionados: (1) Si el oxígeno subió, ¿hasta cuánto subió? (2) ¿Qué causó el incremento? (3) ¿Cuál podría ser la relación entre el oxígeno en ascenso y la evolución de los animales?
FIGURA 10.3 Concentraciones y composiciones isotópicas del molibdeno a lo largo de los últimos 2000 millones de años. Los datos provienen de varias fuentes a través de una compilación cedida cortésmente por Tais Dahl, con datos adicionales de Sahoo y colaboradores (2012).
Tomemos la primera pregunta para empezar. Con independencia de cualquier consideración sobre los animales y sus necesidades de oxígeno, ¿podemos decir algo de los niveles de oxígeno atmosférico del neoproterozoico tardío? Por desgracia, nuestro juego de herramientas no está aún lo bastante desarrollado para aportar una respuesta específica, pero un razonamiento cuidadoso puede ofrecernos unas estimaciones aproximadas. Empecemos con los isótopos del molibdeno. Aunque es cierto que los isótopos del molibdeno apuntan a un incremento en la retirada del molibdeno de los océanos en escenarios marinos óxicos de la era neoproterozoica tardía, la composición isotópica del molibdeno sigue siendo mucho más baja que la que encontramos hoy. Esto indica que, en la época en que emergieron los animales, las condiciones marinas óxicas no eran tan extensas como hoy. Esto implicaría a su vez unos niveles de oxígeno más bajos, quizá incluso mucho más bajos, de los que tenemos en el presente.
Tampoco demasiado bajos, sin embargo. Ya intentamos estimar los niveles de oxígeno atmosférico cuando describimos por primera vez las evidencias de una oxigenación de las aguas profundas en las rocas de la península de Avalon, como vimos más arriba. El argumento era como sigue. En los océanos modernos hay típicamente una zona de mínima concentración de oxígeno que se desarrolla a profundidades desde unos cientos de metros hasta 1500 metros. Este es más o menos el intervalo de profundidades en que vivían los organismos representados por los fósiles de Avalon. El mínimo de oxígeno resulta de la convergencia de dos factores. El primero es el descenso de los organismos de la superficie del océano, que consumen oxígeno al descomponerse. El segundo es el hundimiento del agua superficial fría y rica en oxígeno desde las regiones polares hasta las profundidades del océano. Esta agua forma el agua profunda de los océanos. Muy poca materia orgánica sobrevive el largo tránsito desde la superficie del océano hasta esas profundidades, así que poca descomposición de la materia orgánica tiene lugar, y ello permite al oxígeno persistir. En consecuencia, y al menos en el océano moderno, tenemos altos niveles de oxígeno en la superficie del océano, también altos a grandes profundidades, y más bajos entre medias, lo que genera la zona de mínimo oxígeno. La magnitud de este mínimo de oxígeno varía de un lugar a otro pero, en el mundo moderno, la reducción de la concentración de oxígeno alcanza un mínimo de alrededor de 40 micromoles de oxígeno por litro. En los océanos actuales, las aguas profundas empiezan con una concentración de oxígeno de unos 325 micromoles por litro,
[125] y un mínimo de 40 micromoles por litro en concentración representa el 12% de este valor moderno del agua profunda. Asumimos entonces que el descenso de oxígeno en el lugar donde vivieron los organismos representados por los fósiles de Avalon estaba a un mínimo de unos 40 micromoles por litro (tal vez más). Por último, para dar a los organismos algo de lo que vivir, argumentamos que debía quedar algo de oxígeno después de que esos 40 micromoles por litro se hubieran utilizado. Al final, estimamos que los organismos vivían en un mundo en el que los niveles de oxígeno atmosférico eran del 15% o más de los niveles presentes.
[126] Si hubiera sido menos, la falta de oxígeno habría sofocado a los organismos a la profundidad a la que vivían, aunque unos niveles de oxígeno más altos habrían seguido permitiendo la vida. Así que, si estábamos en lo cierto, el oxígeno era algo así como el 15% o más de los valores modernos durante la era neoproterozoica tardía, pero aun así tenía unos niveles considerablemente menores que los de hoy. Esto es lo mejor que puedo ofrecer con las evidencias disponibles en el presente, pero en el próximo capítulo podremos contrastar esta estimación con otras interpretaciones.
Si hubo un incremento en los niveles de oxígeno atmosférico durante el neoproterozoico tardío, tendrá que haber habido una causa. Hace unos años la respuesta parecía clara. En un importante descubrimiento geoquímico, Andy Knoll aportó en 1986 los primeros resultados detallados de isótopos de carbono de las rocas neoproterozoicas. Estas rocas mostraban una preponderancia de valores enriquecidos en 13C, y si el lector recuerda el capítulo 7, esto significa unas altas tasas de enterramiento de carbono. También significa altas tasas de liberación de oxígeno a la atmósfera. El supercontinente Rodinia estaba rompiéndose en unidades continentales más pequeñas durante el neoproterozoico tardío, y Andy razonó que, con el incremento consiguiente del área de costa,
[127] habría más deposición de sedimentos, produciendo unas tasas elevadas de enterramiento de carbono orgánico. Refinando este argumento, Lou Derry —ahora en Cornell, pero entonces un
posdoc de Harvard— introdujo otros sistemas de isótopos que ayudaron a precisar el máximo de enterramiento de carbono orgánico en unos 580 millones de años atrás.
[128] Esto resultó perfecto. Problema resuelto.
Como ocurre a menudo en ciencia, sin embargo, una idea interesante se somete a pruebas y a la acumulación de más datos y, en este caso, un montón más de datos. Mientras que Lou disponía de un centenar de mediciones de isótopos del carbono para construir su modelo, ahora hay miles (figura 10.4). Estos nuevos datos han preservado muchos de los rasgos generales de los isótopos del carbono que modeló Lou, pero la datación ha cambiado, y han emergido nuevos rasgos que confunden el cuadro de manera notable. Uno de ellos es una gran anomalía que conduce a unos valores muy mermados en 13C, conocida como la anomalía isotópica del carbono de Shuram-Wonoka (figura 10.4). La datación de esta anomalía y su duración son inciertas, pero la mayoría coincide en que ocurrió algún tiempo después de la glaciación Glaskiers, que acabó hace 580 millones de años, y antes de 551 millones de años atrás. Actualmente ha sido identificada en muchos lugares alrededor del mundo, y parece ser un rasgo robusto del ciclo del carbono del proterozoico tardío.
[129]El problema con esta anomalía es que resulta casi imposible entender cómo se originó, en nuestros esquemas normales sobre el funcionamiento del ciclo del carbono. La cuestión es de dónde sacar todo ese carbonato mermado en 13C que acaba en la caliza. Hay una variedad de posibles soluciones a este problema, incluyendo una ingeniosa proposición de Dan Rothman, del MIT. Imaginó que, en esa fase de la historia, el océano era algo así como una sopa orgánica, que contenía enormes concentraciones de materia orgánica disuelta. Los compuestos orgánicos disueltos deberían tener la misma señal isotópica mermada en 13C que las algas, como vimos en capítulos anteriores. Dan argumentó que la oxidación ocasional de esta sopa orgánica produjo una gran cantidad de CO
2 mermado en 13C. De hecho, Dan argumentó que se pudo haber producido el bastante CO
2 mermado en 13C para producir la anomalía de Shuram-Wonoka. Es una idea brillante, pero nunca llegué a comprender cómo arrancó ese episodio de oxidación. Por esta y otras razones, Christian Bjerrum y yo propusimos otra solución que implicaba la oxidación de un vasto reservorio de metano (que contenía incluso más carbono mermado en 13C que los compuestos orgánicos disueltos), sobre el que puedes aprender más siguiendo la nota 14.
[130] En cualquier caso, entender la anomalía de Shuram-Wonoka requiere un poco de pensamiento lateral, pero todas estas soluciones tienen un precio, al menos en términos de oxígeno. Así que, ya estemos hablando de compuestos orgánicos disueltos o de metano para formar la anomalía de 13C de Shuram-Wonoka, se requiere una gran cantidad de oxígeno para ejecutar la oxidación,
[131] resultando en un enorme sumidero de oxígeno.
Veamos en qué punto estamos. Las evidencias geoquímicas indican una oxigenación incrementada de los océanos hace unos 580 millones de años, tal vez algo antes, y en concierto aproximado con la expansión de la fauna de Ediacara. Tal vez, como insinuaron Andy Knoll y Lou Derry, fue disparada por unas tasas altas de enterramiento de materia orgánica asociadas con la fragmentación del supercontinente Rodinia. Poco después de esto, sin embargo, la anomalía de Shuram-Wonoka indica un gran sumidero de oxígeno y la posibilidad de una significativa declinación del oxígeno en la atmósfera y los océanos. No sabemos cuánto puede haber bajado el oxígeno por esta anomalía, pero parece que no se redujo hasta el punto de afectar la respiración de los animales primitivos.
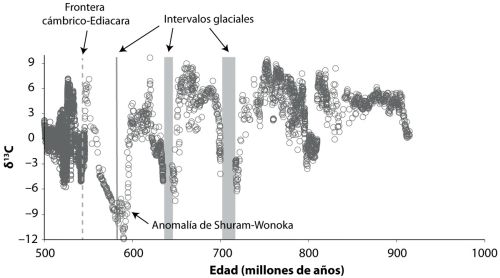
Figura 10.4 Registro neoproterozoico de la distribución de isótopos del carbono inorgánico. Se indica la anomalía de Shuram-Wonaka, así como los principales episodios de glaciación. La relación entre la anomalía de Shuram-Wonaka y la glaciación (que podría muy bien haber ocurrido después) y la duración de la anomalía son muy inciertas. Datos compilados y cedidos cortésmente por Galen Halverson, de la Universidad McGill, y Matt Saltzman, de la Universidad Estatal de Ohio.
¿Qué ocurrió a continuación? ¿Se elevó el nivel de oxígeno de nuevo hasta niveles pre-Shuram-Wonoka? Parte de las evidencias geoquímicas esbozadas más arriba podrían indicarlo, pero el registro de isótopos de carbono no revela gran cosa. De hecho, el modelo de Lou sólo mostraba un incremento transitorio de la liberación de oxígeno hace unos 580 millones de años. Después de eso, el enterramiento de carbono orgánico y la liberación de oxígeno revirtieron a los niveles de fondo. Además, y para acabar de confundir las cosas, el registro de isótopos de carbono revela otra gran reducción de 13C (figura 10.4) justo en la frontera entre el precámbrico y el cámbrico, coincidiendo con la gran expansión de la vida animal. Tomándolo literalmente, la vida animal se incrementó en magnitud y diversidad cuando la liberación de oxígeno a la atmósfera se desvanecía. Hay muchas cosas aquí que no entendemos.
Bien, ¿y qué hay de los animales? Como se mencionó antes en este capítulo, ha habido el prejuicio, que se remonta a Nursall, de que la evolución de los animales, o al menos de los animales macroscópicos mótiles, fue activada por un incremento en las concentraciones de oxígeno atmosférico. En 1982, de hecho, Bruce Runnegar, de UCLA, intentó poner algunos números en eso. Tomó como ejemplo el fósil de Ediacara Dickensonia, al que veía por entonces como un tipo de gusano anélido. La mayoría estaría ahora probablemente en desacuerdo con esa idea, pero, para el propósito del cálculo, realmente no importa gran cosa lo que fuera Dickensonia. Lo importante es la suposición razonable que hizo Bruce de que Dickensonia obtenía su oxígeno por difusión pasiva a través de su superficie exterior. Con esa suposición, Bruce concluyó que se requerirían unos niveles de oxígeno de al menos el 10% de los actuales para mantener la respiración de Dickensonia. Ignorando las posibles acrobacias del oxígeno asociadas con la anomalía de Shuram-Wonoka, la demanda de oxígeno para Dickensonia parece encajar con nuestros argumentos geoquímicos que indican que, hace unos 580 millones de años, el oxígeno aumentó hasta al menos el 15% del nivel presente. Suena bastante bien. En este escenario, la evolución de los animales grandes y posiblemente motiles como Dickensonia se hizo posible por un aumento del oxígeno hasta unos niveles propicios para su subsistencia. En palabras de Andy Knoll, los animales motiles evolucionaron en un «entorno permisivo».
Este tipo de lógica hace encogerse a algunos paleontólogos, y en concreto a Nick Butterfield, de la Universidad de Cambridge. Casi le puedo ver estremecerse cada vez que lee sobre una posible relación entre la concentración de oxígeno y la evolución animal, sobre todo si el argumento viene de un geoquímico. El punto de vista de Nick es que el oxígeno debería dejarse fuera de la ecuación. Argumenta que los propios animales han construido las condiciones de cambio ambiental, y que cualquier desarrollo evolutivo que podamos atribuir a un «entorno permisivo» fue hecho por los propios animales. Nick escribió en un artículo de 2011:
Facilitando y forzando la diversificación de, por ejemplo, el fitoplancton eucariótico, un cuerpo grande, la bioturbación y la biomineralización, los animales primitivos reinventaron el intercambio químico entre la biosfera y el planeta. Bajo esta luz, las perturbaciones biogeoquímicas del intervalo Ediacara-Cámbrico se ven más probablemente como consecuencias de arriba debajo de la evolución animal que como su causa de abajo a arriba.
Quizá le sorprenda a Nick leer que a mí me gusta mucho esa idea. No cabe la menor duda de que los propios animales (incluyendo el obvio impacto reciente de los humanos) han influido de una forma fundamental en el reciclado biogeoquímico de los elementos y la química de los océanos. Bajo este ángulo, la oxigenación que vemos hace 580 millones de años, o quizás un poco antes, puede reinterpretarse mejor como una redistribución del oxígeno en los océanos debido a la actividad animal, y no como un incremento en los niveles del oxígeno atmosférico. Esta idea, en realidad, se remonta a Graham Logan, John Hayes, Glenn Hieshima y Roger Summons; argumentaron en 1995 que la evolución de un minúsculo plancton animal, el llamado zooplancton, cambió completamente el ciclo del carbono y la distribución de oxígeno en los océanos. La idea es que el zooplancton produce bolitas fecales que se hunden deprisa. Estas se descompondrían menos en las capas altas del océano mientras se hunden hacia el fondo del mar, en comparación con la biomasa microbiana, más pequeña y de hundimiento más lento, que previamente había dominado los océanos.
Puedes pensar en esto de la forma siguiente. Imagina que tienes un cubo con un agujerito en el fondo que deja salir el agua. Si te asomas a un balcón alto y vas bajando el cubo lentamente, perderás mucha agua mientras se aproxima al suelo, y si lo bajas lo bastante lento puedes perder toda el agua mientras el cubo está todavía en el aire. Pero bájalo más deprisa y se perderá mucha menos agua del cubo mientras viaja hacia el suelo. Bien, la pérdida de agua de nuestro cubo es análoga al oxígeno utilizado durante la descomposición de la materia orgánica mientras se va posando desde las capas altas del océano hacia las profundidades. Si la materia orgánica se posa deprisa, se usa menos oxígeno durante su descomposición en las porciones más superficiales del océano (por analogía, menos agua se pierde del cubo) en comparación con la situación en que la materia orgánica se posa despacio. En este segundo caso, mucho más oxígeno se usará en las zonas más superficiales del océano mientras la materia orgánica se posa gradualmente y se descompone.
Cabe por lo tanto imaginar que, con un incremento en la velocidad de hundimiento de la materia orgánica, se usará menos oxígeno en su descomposición en las capas altas del océano (lo que significa en los cientos de metros superiores). Esto resultaría en un incremento de la oxigenación de esas aguas en comparación con la situación en que la materia orgánica se posa despacio. En verdad, un incremento en la oxigenación de los océanos es lo que nos dicen nuestras aproximaciones químicas, pero, si seguimos la lógica que acabamos de esbozar, ese incremento podría muy bien ocurrir sin ningún cambio en los niveles de oxígeno atmosférico. De ser así, ello podría ayudar a explicar nuestras dificultades para reconocer las fuerzas conductoras de un incremento del oxígeno atmosférico en el neoproterozoico tardío. Puede ser, de hecho, que no hubiera ningún incremento.
Graham Logan y sus colegas ofrecieron también algunos apoyos para su idea de que la evolución animal pudo haber cambiado la dinámica del carbono en los océanos. Examinaron una serie de biomarcadores orgánicos y hallaron muy pocas evidencias de preservación de materia orgánica producida por algas en los organismos fotosintéticos de la parte superior de la columna de agua en sedimentos más antiguos de 590 millones de años. Sin embargo, en sedimentos de 530 millones de años o más recientes, sí encontraron esos restos de biomarcadores. Hay un agujero grande e importante en los datos entre 590 y 530 millones de años atrás, pero Logan y sus colegas arguyeron pese a ello que, antes de 590 millones de años atrás, los organismos fotosintéticos se descomponían en su mayor parte durante su lento descenso hacia el fondo marino. Hace 530 millones de años, en contraste, el zooplancton y otros animales pelágicos surgieron en el ecosistema marino y generaron bolitas fecales que se hundían con rapidez; estas transportaban los restos del fitoplancton superficial al fondo del mar, donde fueron descubiertos por los geoquímicos 500 millones de años después.
Esta es una hipótesis atractiva, pero sigue necesitando pruebas. La historia de la evolución del zooplancton oscila entre lo pobre y lo inexistente. Unos organismos tan pequeños dejan un registro fósil de cuarta fila. Nick Butterfield arguye, sin embargo, que aunque su registro fósil sea malo sigue siendo posible buscar pruebas de su presencia centrándose en lo que podrían haber comido. En este caso, hay un registro abundante de potenciales alimentos, los llamados acritarcos, que supuestamente representan las cubiertas preservadas de algas primitivas o sus equivalentes en estado quiescente. Estos experimentaron un incremento drástico de ornamentación hace unos 630 millones de años que, en opinión de Nick, representa una defensa contra la prelación por el zooplancton recién evolucionado. Este calendario no encajaría muy bien con los resultados de Logan. Recordemos que él y sus colegas argumentaban que la influencia de los animales en el ciclo del carbono vino más tarde, hace unos 590 millones de años o después.
Claramente, nuestro entendimiento de la relación entre el oxígeno y los animales es neblinoso. Hay que esclarecer muchos detalles. Entre otras cosas, tenemos que explorar en detalle cómo un ciclo del carbono cambiante, conducido por la evolución animal, afectaría a la oxigenación del océano y a su registro, que pueden revelarse con muchos indicadores indirectos diferentes. Esto va a requerir el diseño de modelos oceánicos, con toda probabilidad. Parece una posibilidad real, sin embargo, que la historia de la oxigenación del océano y su relación con la evolución animal tenga al menos tanto que ver con lo que estaba pasando en los océanos como con lo que estaba pasando en la atmósfera. Y, de ser esto cierto, sería igualmente posible que los animales motiles evolucionaran en un entorno que ya había sido «permisivo» durante algún tiempo antes de su aparición.
[132] Será muy interesante ver cómo se desarrolla esta historia.
Capítulo 11
Oxígeno fanerozoico
Solicité plaza en un total de cinco escuelas de postgrado, y cuatro de ellas estaban en el noreste de Estados Unidos: la Universidad de Columbia, la Universidad de Yale, el Instituto Oceanográfico de Woods Hole y la Universidad de Rhode Island. Me imaginé que podría visitarlas todas si me tomaba unos diez días para ello. Así que empaqueté mi furgoneta Volkswagen con un saco de dormir y unas cuantas cosas para comer y beber y emprendí el viaje desde mi apartamento en Oxford, Ohio. En Columbia, o más exactamente en el Observatorio Geológico Lamont-Doherty (ahora llamado Observatorio Terrestre Lamont-Doherty), estaba citado para entrevistarme con el eminente oceanógrafo Wally Broecker. Algo que no puedo recordar obligó a Wally a salir de la ciudad justo el día que yo llegué, pero pasé un día maravilloso con su buena colega Tara Takahashi y todo un plantel de impresionantes estudiantes y científicos de plantilla. Mi siguiente parada era Yale, donde tenía previsto verme con Bob Berner. Tras llegar a los Laboratorios de Geología Kline, una monstruosidad de hormigón, sin ventanas ni alma,
[133] diseñada por un arquitecto de fama mundial (el fallecido Philip Johnson), me condujo al despacho de Bob Berner. Allí, un hombre alto con un polo de cuello alto azul y una amplia sonrisa saltó de su escritorio y me saludó con un apretón de manos. Bob me dedicó mucho tiempo. Me enseñó los laboratorios, me llevó a comer y me explicó en detalle todos los proyectos en marcha del laboratorio. Me presentó a Rob Raiswell, una experiencia de las que te cambian la vida (véase la nota 7 del capítulo 9). También me explicó que me había perdido por muy poco a Bob Garrels, que visitaba Yale tres meses al año. Bob Garrels (a quien conocimos en el capítulo 5) era uno de mis héroes científicos incluso en esa época, antes de que yo me sumergiera en la geoquímica. El día acabó con una charla con los estudiantes de postgrado Bernie Boudreau y Mike Velbel, que me aseguraron que New Haven era un sitio espantoso para vivir y que, teniendo elección, sería una locura venir a Yale.
Antes de buscar una escuela de postgrado, pasé dos años trabajando con Bill Green, de la Universidad de Miami en Ohio, sobre la química del lago Vanda, un lago sulfuroso permanentemente estratificado en el valle McMurdo de la Antártida. Mi trabajo incluía tres meses en los hielos antárticos, y el resto del tiempo en el laboratorio; por lo demás vivía sin preocupaciones, sin ataduras y pasándomelo como en mi vida. Por mi trabajo en el lago Vanda, me di cuenta de que quería investigar el papel de los procesos microbianos para dar forma a la química del entorno. Bob era un experto muy conocido en esta área, y los proyectos que describió me interesaron muchísimo. También me explicó, muy animado, unos nuevos modelos que estaba haciendo con Bob Garrels. Explicó algo sobre una justificación cuantitativa de la historia de las concentraciones del CO2 atmosférico a través del tiempo, controladas en gran medida por procesos geológicos. No lo entendí muy bien entonces, pero el entusiasmo de Bob lo hacía parecer importante. Mal podía yo saber entonces que los dos Bobs (Berner y Garrels) estaban inaugurando una forma enteramente nueva de entender cómo los diversos componentes del sistema terrestre interactúan para regular la química de la atmósfera y los océanos. Esto era pensar a lo grande en su máxima expresión.
Unos meses después de mi viaje, las cartas empezaron a llegar: rechazo de la Universidad de Washington; aceptación de la Universidad de Rhode Island; rechazo de Woods Hole; y por último, aceptaciones de Yale y Columbia. Esto planteaba un dilema. Estaba profundamente impresionado por Bob y su estrategia de investigación, pero Wally Broecker era (y es) uno de los investigadores del clima más creativos e influyentes en activo, y el Observatorio Terrestre Lamont-Doherty es una de las mejores instituciones oceanográficas del mundo. La decisión era difícil, pero mi corazón decía Yale, así que allí me fui.
Mi tesis evolucionó hacia un estudio de los sedimentos marinos modernos, y en concreto del papel de los procesos microbianos, dirigidos tanto por el hierro como por el azufre, en el control de la química de los sedimentos y la preservación del carbono orgánico en los sedimentos. Se trataba de un estudio, de campo y de laboratorio, que implicaba la recolección, loncheado, troceado y análisis de diversos fangos marinos. Bob, por otra parte, que llevaba mucho tiempo sin trabajar en el laboratorio, se ocupaba a una escala mucho más ambiciosa intentando conceptualizar cómo funcionaba el sistema Tierra. No mucha gente puede hacer esto.
[134] Requiere la habilidad especial de extraer las verdades simples de unos comportamientos complejos, y de figurarse cómo esas verdades se interconectan de forma cuantitativa. También se precisa una increíble base de conocimiento. Para modelar las concentraciones atmosféricas de CO
2, hay que percibir que debe considerarse una combinación de múltiples procesos. Estos incluyen la erosión en tierra firme, que es influida por diversos parámetros como la temperatura, el nivel del mar (que controla el área continental), el ciclo hidrológico, la elevación continental y el tipo de vegetación terrestre.
[135] Los niveles atmosféricos de CO
2 también vienen controlados por la tasa de entrada de CO
2 desde los volcanes, que está controlada a su vez por la tasa de pérdida de calor del manto, los tipos de sedimento que están reentrando al manto por subducción y la propia tasa de subducción. Y estas son sólo algunas de las consideraciones. Pero Bob (junto a Bob Garrels, y también Tony Lasaga) resolvió, primero, qué procesos del sistema terrestre deben ser considerados como controles del CO
2 atmosférico y, segundo, cómo modelarlos.
Tuve la suerte de sentarme en la banda lateral durante todo este increíble ejercicio intelectual. Recuerdo haber pensado en lo atrevido —aunque apasionante— que resultaba poner números concretos a la historia de la evolución atmosférica; esta es una historia perdida hace mucho para la medición directa (¡qué útil resultaría esa máquina del tiempo!), pero que ahora se puede imprimir en un gráfico y tener en la mano. Me habría gustado ser parte de ello, pero carecía de los conocimientos y habilidades necesarias. Bob, además, insistía en que esos no eran proyectos para estudiantes. Eran de largo demasiado difíciles y demasiado arriesgados para eso.
Hacia el final de mi tesis, sin embargo, sí que tuve la oportunidad de implicarme un poco. En este punto, Bob estaba intentando modelar la historia del oxígeno atmosférico. El modelado del oxígeno partía del marco intelectual establecido durante el modelado del CO2 atmosférico, pero con sustanciales contribuciones de otros lugares también. Durante ese proceso hubo una fermentación de ideas sobre la forma en que los ciclos del carbono, el azufre y otros elementos podrían conectarse y entenderse cuantitativamente. De hecho, en muchos de los congresos geológicos internacionales, los principales jugadores de este campo mantenían «carreras de ciclos» en las cafeterías de los hoteles, donde intercambiaban ideas alimentados por cantidades generosas de pizza y cerveza. Bob Garrels era la figura paterna de esta gente, y el modelado del oxígeno de Bob Berner se benefició de muchas contribuciones de Garrels, así como de Abe Lerman, de la Universidad Northwestern, Fred Mackenzie, de la Universidad de Hawai, y Lee Kump, uno de últimos estudiantes de doctorado de Bob Garrels y ahora en la Universidad Penn State.
De hecho, antes de que Bob Berner empezara a modelar el oxígeno atmosférico, Lee Kump y Bob Garrels publicaron un intento previo de modelo, que produjo una historia de las concentraciones de oxígeno atmosférico durante los últimos 100 millones de años de historia de la Tierra. Esto es un trabajo maravilloso en el que las tasas de liberación de oxígeno a la atmósfera se cuantificaron a partir del registro de carbono y azufre (como exploramos en los capítulos 8 y 9), y donde se derivaron unas ecuaciones para cuantificar las tasas de retirada de oxígeno durante la erosión de las rocas que contienen carbono o azufre. La concentración de oxígeno en la atmósfera resultaba del equilibrio cinético entre las tasas de producción y consumo de oxígeno. En general, Kump y Garrels hallaron que las concentraciones de oxígeno eran elevadas cuando el registro de isótopos indicaba unas altas tasas de liberación de oxígeno a la atmósfera, y más bajas cuando el registro de isótopos indicaba unas tasas bajas de liberación. Esto quizá no sea muy sorprendente, pero el desafío, bellamente aceptado por Kump y Garrels, era averiguar cómo todos esos procesos se vinculaban cuantitativamente, y cómo las concentraciones de oxígeno podían extraerse de los datos isotópicos.
De hecho, la primera entrada de Bob en el modelado de oxígeno se apoyó con firmeza en este trabajo, pero añadió un importante componente adicional, que es la idea de reciclado rápido. Esta idea se discutió en el capítulo 5, y su punto básico es que los sedimentos depositados más recientemente serán también los más propensos a la erosión mediante procesos como cambios en el nivel del mar o la elevación de la tierra. Así, a través del reciclado rápido, unas altas tasas de producción de oxígeno por enterramiento de sedimentos ricos en materia orgánica conducirán deprisa a unas altas tasas de consumo de oxígeno por exposición de esos sedimentos orgánicos a la erosión. Desde la perspectiva del modelado, y como vimos en el capítulo 5, el reciclado rápido (junto con varias retroalimentaciones negativas) ayuda a amortiguar las variaciones de oxígeno. Esto es importante porque los flujos de oxígeno a través de la atmósfera durante el enterramiento del carbono orgánico y la pirita, y por erosión, son enormes en comparación con la cantidad relativamente baja de oxígeno en la atmósfera. Así, todo el oxígeno en la atmósfera actual se recicla a través de procesos geológicos de liberación de oxígeno (enterramiento de carbono orgánico y pirita) y de consumo de oxígeno (erosión) en el plazo de dos o tres millones de años. Esto te puede parecer mucho, pero no lo es a escalas de tiempo geológico, y pequeños desequilibrios entre las tasas de liberación y consumo de oxígeno se traducen enseguida en cambios rápidos de la concentración de oxígeno atmosférico.
Además del reciclado rápido, Bob Berner tuvo otra gran idea. Razonó puede haber formas más directas que los registros de isótopos para obtener una historia del enterramiento de carbono y el azufre. Para ver cómo funciona esto, empecemos por Aleksandr Borisovich Ronov, un famoso geólogo ruso.
[136] Pasó gran parte de su carrera compilando cuidadosamente mapas geológicos de un grupo de rocas conocido como la plataforma rusa, y de otras rocas por todo el mundo. Ronov utilizó estos mapas para evaluar el volumen de los diferentes tipos de rocas preservadas a través del tiempo geológico. De este modo, Ronov determinó, entre otras cosas, el volumen de sedimentos preservados de entornos marinos, de entornos continentales y de depósitos de carbón. Este conjunto de datos (y gran parte de los otros datos que compiló Renov) son una verdadera mina de oro de información, y en la figura 11.1 se recoge un gráfico que muestra la distribución de diferentes tipos de rocas a través del eón fanerozoico.
Bob Berner percibió que cada uno de esos diferentes tipos de sedimentos portaba, como promedio, diferentes cantidades de carbono orgánico y azufre pirítico. Por ejemplo, los depósitos de carbón son ricos en carbono orgánico y por lo general pobres en azufre,
[137] mientras que otros sedimentos depositados en los continentes (a menudo compuestos de arenas y gravilla) son pobres tanto en carbono orgánico como en azufre pirítico. Los sedimentos marinos tienen unos niveles intermedios de carbono orgánico y unos contenidos elevados de azufre pirítico en comparación con los depósitos terrestres. Todo esto significa que, si conocemos los contenidos medios de azufre y carbono orgánico de estas rocas, así como sus tasas de deposición a través del tiempo, entonces las tasas de liberación de oxígeno a la atmósfera pueden determinarse directamente. Bob asumió que la distribución relativa de los tipos de rocas preservados a través del tiempo, tal y como la revelaron las compilaciones de Ronov, reflejaba su distribución relativa en la época en que se depositaron. Bob también usó diversos escenarios para calcular las tasas de deposición de sedimentos. Estos escenarios variaban desde tasas constantes de deposición total de sedimentos a través del tiempo hasta tasas variables,
[138] pero al final la variabilidad en las tasas totales de deposición de sedimentos reveló una escasa influencia en el resultado del modelo.
[139]Así, con estimaciones de los contenidos de carbono y azufre en los diferentes tipos de sedimento, y con una tasa constante de deposición de sedimentos, Bob calculó las tasas de enterramiento de carbono orgánico y azufre pirítico a través del tiempo. Necesitamos un pequeño detalle adicional. Tras unas cuantas operaciones iniciales del modelo, parecía que algo de retroalimentación en el ciclo del azufre sería importante. Ese fue mi trabajo, y mi modesta contribución al estudio. Hallé que una dependencia del oxígeno en el enterramiento del azufre en los sedimentos marinos sería razonable. Exploramos esta teoría en el capítulo 5, y la idea básica es que, a medida que baja la concentración de oxígeno atmosférico, debería darse una extensión de las condiciones anóxicas y sulfurosas en los océanos. Esto conduce a un incremento en la tasa de enterramiento de pirita y, por lo tanto, en la tasa de liberación de oxígeno a la atmósfera. El efecto es una retroalimentación negativa: unas tasas reforzadas de enterramiento de azufre en condiciones de bajo oxígeno evita que el oxígeno baje demasiado.
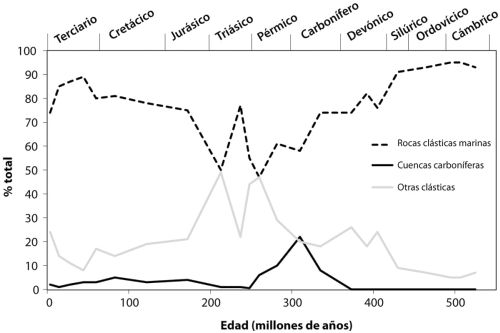
FIGURA 11.1 Distribución de los tipos de sedimentos a lo largo del eón fanerozoico. Datos de A. B. Ronov resumidos en Berner y Canfield (1989).
Para hacerse una idea de los resultados del modelo, en la figura 11.2 se muestran las tasas de enterramiento de carbono orgánico calculadas a partir de las tendencias de abundancia de sedimentos. Y se comparan con las tasas de enterramiento de carbono orgánico calculadas a partir del modelado de los isótopos de carbono, como hizo Bob en su primer estudio. Al menos para mí, hay una similitud extraordinaria entre los resultados de esos dos cálculos. Esto te hace pensar que, al menos durante el eón fanerozoico, tanto el registro geológico de tipos de sedimento preservados como el registro de isótopos de carbono nos están diciendo algo similar y fundamental sobre el ciclo del carbono.
[140] Esto quizá nos pone en una buena posición para decir algo importante sobre la evolución de las concentraciones de oxígeno a lo largo del eón fanerozoico.
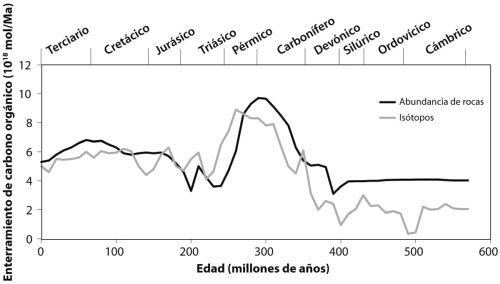
FIGURA 11.2 Tasas de enterramiento del carbono orgánico calculadas tanto por los datos de abundancia de rocas como por los de isótopos. Ma = Mega-años, o millones de años. Redibujado a partir de Berner y Canfield (1989), utilizando la escala de tiempo conocida en el momento de su publicación; la mayoría de las fronteras entre periodos han cambiado un poco desde entonces (véase el prefacio).
Esto también nos lleva a los resultados del modelo, que se muestran en la figura 11.3. El área gris en el gráfico indica la mejor estimación de Bob para el intervalo de concentraciones de oxígeno, según revelan varios análisis de sensibilidad, mientras que la línea representa el mejor resultado del modelo, desde el punto de vista de Bob. El modelo muestra claramente variaciones en el contenido de oxígeno atmosférico. Los análisis de sensibilidad revelaron que el reciclado rápido era importante para amortiguar las fluctuaciones del contenido de oxígeno de la atmósfera, y también que el contenido de materia orgánica (en particular) y el contenido de azufre pirítico (de menor relevancia) de los diversos tipos de sedimento importaban mucho. Por el contrario, el área continental disponible para la erosión era poco importante, como también lo era la tasa total de sedimentación, como apunté más arriba.
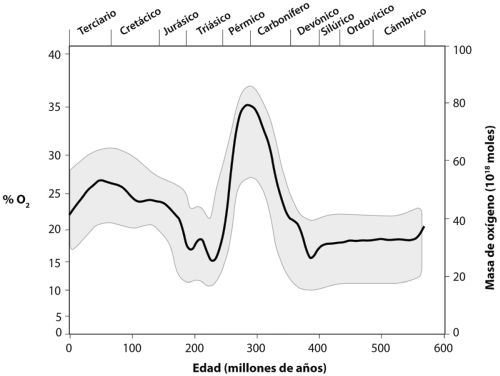
FIGURA 11.3 Concentraciones de oxígeno a lo largo del eón fanerozoico calculadas a partir de los datos de abundancia de rocas. Redibujado a partir de Berner y Canfield (1989), utilizando la escala de tiempo conocida en el momento de su publicación.
Lo más importante de todo, sin embargo, era el tipo de sedimento depositado. El rasgo más obvio del gráfico es la gran anomalía positiva de oxígeno que se ve en los periodos carbonífero y pérmico. Este incremento de oxígeno se disparó por la deposición masiva de carbono orgánico en depósitos de carbón. Esto, por supuesto, ocurrió por una razón o, más probablemente, por un par de razones. La primera es que las plantas terrestres, que se originaron probablemente en el periodo ordovícico temprano, se diversificaron durante el periodo silúrico (véase el prefacio para la escala de tiempo) y, hacia el periodo carbonífero temprano, habían crecido de tamaño y se habían expandido mucho por los continentes. Para alcanzar buena altura, las plantas desarrollaron una serie de moléculas orgánicas duras, como la lignina y la celulosa, que tienden a resistir la descomposición por microbios, sobre todo cuando se acumulan en entornos libres de oxígeno como los sedimentos. Por tanto, la evolución de las plantas tuvo un papel. Y los detalles de la paleogeografía probablemente fueron también importantes. Durante los periodos carbonífero y pérmico, vastas extensiones de ciénagas y tierras pantanosas recolectaron y enterraron cantidades masivas de residuos vegetales orgánicos, y por eso se formó tanto carbón en aquel tiempo. Hoy hay entornos comparables en las Everglades de Florida, y en las turberas que se encuentran en muchos lugares del mundo, sobre todo a altas latitudes.
La caída de la concentración de oxígeno que siguió se debió probablemente a un cambio de paleogeografía y de los tipos de sedimento depositados. Empezando en el periodo pérmico, pero continuando hasta el triásico, se ensambló un supercontinente llamado Pangea. Parece que, como resultado de este ensamblaje, la mayoría de las áreas pantanosas dejaron de estar disponibles. Además, debido a su enormidad, una menor proporción de la lluvia que caía sobre Pangea fluía hacia el mar, y una mayor proporción fluía dentro del propio continente, formando vastas extensiones de depósitos de arcillas arenosas bien drenadas (encontramos arcillas de ese tipo en el capítulo 8). Estos sedimentos continentales arenosos carecen virtualmente por completo de materia orgánica, y por tanto su formación no aporta ninguna fuente de oxígeno a la atmósfera. Por tanto, en el modelo de Bob, el cambio a una gran deposición de arcillas rojas durante los posteriores periodos pérmico y triásico redujo el aporte de oxígeno a la atmósfera, lo que resultó en un descenso brusco de la concentración de oxígeno, quizá a niveles significativamente inferiores a los de la atmósfera actual.
De este modo, los cambios en el tipo de sedimento depositado, que vienen controlados por una combinación de factores como la evolución de las plantas, la paleogeografía y el clima, parecen haber controlado la tasa de entrada de oxígeno en la atmósfera, ejerciendo una influencia esencial en los niveles de oxígeno atmosférico. Incluso puede haber algunas evidencias que respalden esta historia, o al menos parte de ella. Es bien sabido que los insectos gigantes formaban parte de la biosfera del carbonífero y el pérmico temprano; algunos de ellos tenían un tamaño casi de pesadilla. Hay que imaginárselo, escolopendras de más de un metro de longitud, arañas gigantes con patas del ancho de una silla de despacho (unos 50 centímetros) y libélulas con 70 centímetros de ala. De hecho, estas libélulas gigantes movieron hace más de un siglo a los paleontólogos franceses Harlé y Harlé a proponer que los niveles de oxígeno atmosférico del periodo carbonífero debieron ser más altos que los actuales. Parte de su razonamiento era que una mayor presión atmosférica, causada por mayores niveles de oxígeno, ayudaría a mantener en el aire a estos masivos aviadores. Además, y tal vez de forma más importante, razonaron que una mayor disponibilidad de oxígeno permitiría las mayores tasas de respiración que unos organismos tan grandes requerirían para volar, sobre todo si tenemos en cuenta que obtienen su oxígeno por difusión a través de sus sistemas traqueales.
Estas ideas, aunque son evidentemente razonables, han espoleado debates masivos, y también un cuerpo creciente de estudios experimentales. Nick Butterfield (a quien conocimos en el capítulo 10), siempre llevando la contraria, ha hecho su intento con las libélulas. Cree que las consideraciones ecológicas han sido un impulso mucho más fuerte hacia el gigantismo, relegando cualquier ayuda de unos elevados niveles de oxígeno a un papel secundario. O se puede exagerar la importancia de la ecología como conductora de la evolución, pero Nick no considera el gigantismo contemporáneo en los grupos de insectos no voladores. Seguramente estarán expuestos a diferentes condiciones ecológicas que las libélulas. Así que, ¿es una mera coincidencia que un conjunto distinto de interacciones ecológicas conduzca también al gigantismo en los grupos de insectos sin vuelo? ¿O es que el conductor es otro, como el oxígeno?
Ha habido también una serie de experimentos directos en varios grupos de insectos, pero han conducido a resultados equívocos. Gran parte de este trabajo es obra de Jon Harrison y su grupo de la Universidad Estatal de Arizona. Cuando el grupo de Jon crio diferentes tipos de insectos a bajos niveles de oxígeno, se observó en general una reducción del tamaño corporal, un crecimiento más lento y unas menores tasas de supervivencia (volveremos a este asunto más abajo). Sin embargo, cuando se criaron con niveles de oxígeno mayores que los actuales, los resultados fueron contradictorios. En algunos casos, como con los escarabajos, las moscas del vinagre y las libélulas, el tamaño corporal se incrementó a niveles altos de oxígeno. En el gusano de la harina gigante Zophobus morio, el tamaño del cuerpo crece a niveles de oxígeno moderadamente altos, pero empieza a menguar de nuevo si se sigue subiendo el oxígeno. En muchas otras especies no hubo ningún efecto.
En suma, aunque los resultados fueron variados, algunos experimentos mostraron claramente un incremento del tamaño corporal a elevados niveles de oxígeno. Este resultado fue probablemente una respuesta fisiológica directa a las altas concentraciones de oxígeno, aunque los organismos más grandes podrían también haber sido seleccionados en experimentos que prosiguieran durante múltiples generaciones. En cualquier caso, la escala de tiempo del experimento es probablemente demasiado corta para permitir los lentos cambios evolutivos que seguramente resultaron en un verdadero gigantismo en el registro fósil. Por tanto la cuestión sigue abierta, pero una relación causal entre los niveles elevados de oxígeno y el gigantismo de los insectos en el carbonífero y principios del pérmico sigue siendo una explicación plausible.
Volvamos a los modelos. Desde que publicamos la historia de los niveles fanerozoicos de oxígeno a partir de los datos de abundancia de rocas, Bob ha vuelto al modelado de oxígeno basado en los registros de isótopos de carbono y azufre. Este modelo se llama Geocarbsulf, y es un gran modelo de las historias del CO2 y del O2 atmosféricos. Los resultados del modelo para el oxígeno se presentan en la figura 11.4, y son muy similares a nuestro modelo previo (compárese con la figura 11.3), lo que refuerza la idea de que la abundancia de rocas y los datos de isótopos revelan un cuadro similar de los procesos que controlan la liberación de oxígeno a la atmósfera.
Pero aquí no acaba la historia. Tim Lenton, de la Universidad de Exeter, su director de tesis Andy Watson, de la Universidad de East Anglia, y su estudiante de doctorado Noam Bergman también intentaron modelar el oxígeno atmosférico (y el CO
2) durante el fanerozoico, pero con un enfoque algo distinto. Su modelo, llamado COPSE (iniciales de carbono, oxígeno, fósforo (P), azufre (S) más la E de evolución), se apoya en la filosofía de la hipótesis de Gaia de James Lovelock.
[141] No entraré aquí en los detalles, pero, en su forma más básica, la hipótesis de Gaia postula que los organismos han tenido un papel activo en el moldeado de la química del entorno. Esta idea estaba implícita en el modelo que hicimos previamente Bob y yo, puesto que los organismos, y en particular la evolución de las plantas terrestres, tuvieron un papel principal al influir la abundancia de carbono orgánico en las rocas, que a su vez alteró los niveles de oxígeno. Los organismos han tenido una presencia más explícita en los modelos recientes de isótopos, ya que influyen directamente en el ciclo del carbono de formas diversas.
[142]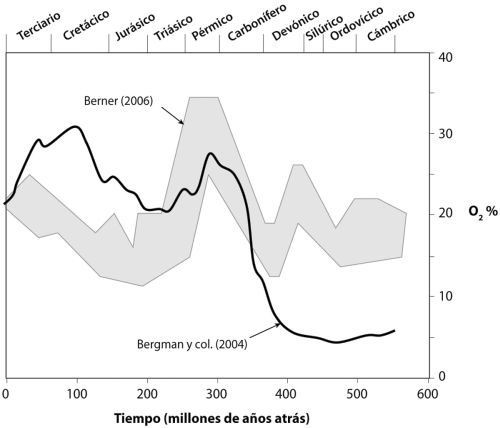
FIGURA 11.4 Resultados del modelo Geocarbsulf de Berner comparados con los del modelo COPSE de Bergman y otros. El área gris alrededor de los resultados del modelo de Berner abarca el intervalo de incertidumbre estimado por Berner, así como resultados más recientes que se han presentado como refinamientos del modelo Geocarbsulf original.
En vez de basarse en los isótopos de carbono y azufre para derivar de un modo fundamental las tasas de producción de oxígeno, el modelo COPSE se alimenta de varios factores externos, de naturaleza tanto geológica como biológica. Los factores incluyen las tasas de emisión de gases volcánicos, las tasas de elevación tectónica, la evolución de las plantas terrestres, el estímulo de las plantas terrestres sobre la erosión, la localización del enterramiento de carbono orgánico en los océanos (profundos o someros) y el incremento de la luminosidad solar durante el eón fanerozoico.
[143] En realidad, muchos de esos factores pueden encontrase en los modelos de Bob, pero el modelo COPSE difiere significativamente de los de Bob en algunas de sus retroalimentaciones. Por ejemplo, el modelo COPSE impone una sensibilidad al oxígeno en las tasas de oxidación durante la erosión de los sulfuros y el carbono orgánico en tierra firme. Los modelos iniciales de Kump y Garrels incorporaron esa retroalimentación (recordemos la discusión entre Karl Turekian y Bob Garrels en el capítulo 5), pero Bob la retiró de sus modelos hace mucho. El modelo COPSE incorpora también una retroalimentación por incendios forestales (que también exploramos en el capítulo 5) para evitar que las concentraciones de oxígeno atmosférico suban demasiado. El modelo registra también los nutrientes en el océano, principalmente el fósforo, para regular el enterramiento de materia orgánica en los sedimentos. En suma, en vez de usar las curvas de isótopos de carbono y azufre como ejes impulsores, el modelo COPSE está calibrado para intentar obtener esas curvas de una menara tan fiel como sea posible.
[144] Estas curvas son una parte integral del registro geológico y, si no se pueden explicar, al menos a trazos gruesos, es que algo hay sospechoso en el modelo.
Los resultados del modelo COPSE se muestran junto al modelo Carbsulf en la figura 11.4. Los dos modelos tienen fuertes similitudes, sobre todo en el incremento de concentración de oxígeno que acompaña la emergencia de las plantas terrestres y el subsiguiente enterramiento de carbón en las zonas pantanosas, como discutimos antes, pero hay también diferencias importantes. La principal es el bajo nivel de oxígeno que produce el modelo COPSE al principio del eón fanerozoico. En el modelo COPSE, este rasgo surge porque la posterior evolución y extensión de las plantas terrestres genera un cambio fundamental en la regulación de oxígeno, dando lugar a niveles de oxígeno más altos. Así, en el modelo COPSE, unos niveles relativamente bajos de oxígeno atmosférico son el estado estable antes de la evolución de las plantas terrestres. Si esto es cierto, los bajos niveles de oxígeno que predice el modelo COPSE al principio del eón fanerozoico pueden aportar una estimación de los máximos niveles promedio de oxígeno en la era neoproterozoica tardía, como discutimos en el último capítulo. Por tanto, distinguir entre las predicciones de los modelos COPSE y Geocarbsulf tiene importantes implicaciones para esclarecer las historias del oxígeno atmosférico tanto en el proterozoico tardío como en el fanerozoico temprano.
Tais Dahl y Emma Hammarlund, a quienes encontramos en el último capítulo, pueden haber hallado la forma de distinguir entre los resultados de estos dos modelos.
[145] Recordemos que analizaron la composición isotópica del molibdeno en rocas sedimentarias depositadas en antiguos entornos ricos en sulfuro (euxínicos). Esta estrategia ha sido mencionada en el último capítulo (véase también la nota 14 del capítulo 9), pero, expuesto brevemente, cuanto más alto sea el valor de δ98Mo, más Mo se ha retirado de los océanos en condiciones oxigenadas, y menos se ha retirado en entornos sulfurosos como el mar Negro. Dicho de otro modo, como primera aproximación, cuanto más alto sea el valor de δ98Mo, más oxigenados están los océanos. Lo que encontraron Tais y Emma fue un incremento del valor de δ98Mo hace unos 400 millones de años (figura 10.3). ¿Suena familiar? Desde luego que sí, eso es más o menos el tiempo de la expansión de las plantas terrestres por los continentes, y el tiempo en que el modelo COPSE indica una elevación importante del nivel de oxígeno atmosférico. Esto parece apoyar los resultados del modelo COPSE para el eón fanerozoico temprano.
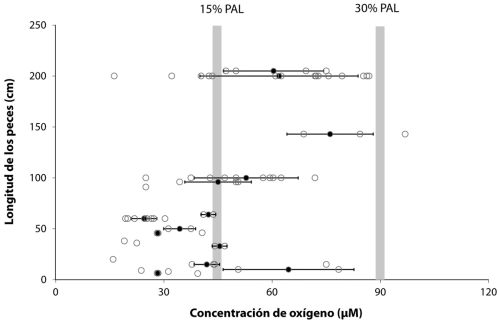
FIGURA 11.5 Relación entre el tamaño de los peces y su supervivencia bajo distintos niveles de oxígeno. El gráfico muestra el llamado LC50, que representa el nivel de oxígeno al que el 50% de la población muere en un tiempo dado. Los promedios y las desviaciones estándar se muestran cuando se estudiaron múltiples ejemplos de la misma especie. Las barras verticales grises corresponden a concentraciones de oxígeno del 15% y el 30% de los niveles atmosféricos actuales (PAL = present atmospheric level, nivel atmosférico presente). Nótese que sólo los peces pequeños sobreviven a niveles de oxígeno inferiores a 15% PAL. Compilación de datos cedida cortésmente por Emma Hammarlund.
Hay más. La subida del valor de δ98Mo también coincide con un cambio bastante profundo del tamaño de los peces en los océanos. En efecto, observamos la emergencia de peces como Dunkleosteus, un auténtico monstruo de hasta 10 metros de largo con un espeso cráneo blindado y unas mandíbulas construidas para triturar. Como discutimos antes con las libélulas, parece razonable que unos peces más grandes y energéticos requirieran más oxígeno, y de hecho esto parece ser cierto para los peces modernos: en promedio, los peces pequeños pueden tolerar menores niveles de oxígeno que los peces grandes (figura 11.5). Se puede observar una pauta similar en la historia de otras criaturas marinas. Por ejemplo, los euriptéridos, o escorpiones de mar, son unos antiguos artrópodos que aparecieron en el periodo ordovícico. Algunos miembros de este grupo crecieron hasta tallas superiores a tres metros cerca de la frontera entre el silúrico y el devónico, hace unos 420 millones de años. Así que se puede argumentar, como hicieron Tais y Emma, que un incremento del nivel de oxígeno atmosférico, asociado a la emergencia de las plantas terrestres, condujo al gigantismo de los peces (y los euriptéridos) en los océanos. Hubo, sin embargo, algunos grandes organismos en los océanos antes de la emergencia de las plantas terrestres. Está, por ejemplo, el Anomalocaris del cámbrico medio, que medía un metro y está bien representado en la Burgess Shale del oeste de Canadá y los depósitos de Chengjiang en China. También el género Megalogratidae alcanzó cerca de un metro en el periodo ordovícico tardío. También hubo ejemplos de nautiloides parecidos a calamares que medían varios metros (del orden Endocerida) en el periodo ordovícico, hace unos 450 millones de años. La presencia de esos gigantes primitivos no implica que Tais y Emma estén equivocadas. Es posible que, aunque estos animales fueran grandes, su estilo de vida fuera más sedentario, con una menor demanda de oxígeno que los peces del mismo tamaño. Una mirada más atenta a la fisiología de las especies nadadoras grandes y pequeñas, rápidas y lentas, será importante para desenmarañar estos asuntos.
Verdaderamente, tenemos aún mucho que aprender. El eón fanerozoico, sin embargo, con su abundante registro fósil y su relativa abundancia de rocas sedimentarias, ofrece la posibilidad de entender la historia de los niveles del oxígeno atmosférico. Esa historia puede ser interrogada desde múltiples ángulos, y parece abarcar una bella relación entre la evolución de las concentraciones de oxígeno atmosférico y la evolución de la vida en la Tierra.
Epílogo
Ha sido un largo viaje. Al final, espero que podamos estar de acuerdo en que la Tierra es un lugar bastante especial. Se halla a una distancia del Sol que nos sitúa en la zona habitable, permitiendo que persista el agua líquida. Esta persistencia se favorece por una regulación activa de la temperatura, que se promueve mediante la relación entre los niveles atmosféricos de CO2, la eyección de CO2 desde el manto y el control térmico de la erosión. La temperatura se controla también por la tectónica de placas; la misma tectónica de placas también dirige el reciclado de materiales crucial para la vida y crucial para la liberación de oxígeno a la atmósfera. Si eliminas la tectónica, todavía se podría hallar agua líquida en la Tierra, al menos en algunos sitios, pero la vida se vería gravemente reducida en abundancia. No habría un suministro continuo de nutrientes para alimentarla. El ciclo de sedimentos no tendría lugar, y la materia orgánica y el azufre pirítico, fuentes últimas de oxígeno en la atmósfera, no se enterrarían. Sin la tectónica, por tanto, incluso si existieran los organismos productores de oxígeno, es improbable que el oxígeno se pudiera acumular en la atmósfera a niveles apreciables. Resulta que la Tierra, tal y como es, tiene lo que hay que tener para ser un planeta acumulador de oxígeno. Cualquier otro planeta en estrellas lejanas que tenga oxígeno en la atmósfera también necesitaría eso mismo que hay que tener, probablemente. Esta es la razón de que la búsqueda de mundos habitables de la NASA se centre en hallar planetas que orbiten en la zona habitable de su estrella.
Tener lo que hay que tener, sin embargo, no es suficiente. Para que el oxígeno se acumule, primero tiene que ser producido, y esto significa que los organismos productores de oxígeno tienen que evolucionar. En la Tierra, la ruta para la evolución de las cianobacterias productoras de oxígeno fue aparentemente algo enrevesada y compleja. Antes de las cianobacterias, hubo al menos dos tipos diferentes de organismos fotosintéticos que no producían oxígeno: los llamados fotótrofos anoxigénicos. Cada uno de estos había desarrollado un fotosistema diferente para convertir la luz en energía celular, y los fotosistemas acoplados de las cianobacterias emergieron como una fusión aparente entre los dos tipos de fotosistema fototrópico. Para hacer la clorofila, las cianobacterias también tomaron prestados, con algunas modificaciones, los sistemas productores de pigmentos de sus ancestros flototróficos anoxigénicos. Más aún, el fundamental complejo de evolución del oxígeno (oxygen-evolving complex, OEC), que utiliza un racimo de manganeso cuádruple, puedo tomarse prestado también, al menos a trazos gruesos, de las enzimas preexistentes que se usaban para convertir el peróxido de hidrógeno en agua y oxígeno. En suma, las cianobacterias no surgieron de la nada: se necesitaron un montón de desarrollos evolutivos antes de que pudieran emerger. El famoso paleontólogo Stephen Jay Gould, de Harvard, gustaba de discutir las contingencias de la evolución; en otras palabras, el grado en que los resultados evolutivos dependen del azar, como un encuentro casual, una mutación casual o la supervivencia casual bajo el estrés ambiental.
La cuestión, entonces, sería: si «pusiéramos la cinta otra vez» (en palabras de Gould), ¿el resultado sería el mismo? ¿Evolucionarían las cianobacterias del mismo modo? ¿Emergerían los fotosistemas acoplados de la misma forma, o emergerían en absoluto, o algo diferente habría ocupado su lugar? ¿Y qué hay del racimo de manganeso? Puede que estas cuestiones sean más filosóficas que científicas, pero no son enteramente triviales. En la ciencia ficción nos sacamos de la manga mundos con aire respirable, y el oxígeno es uno de los parámetros de búsqueda que los científicos usan para rastrear planetas habitables (o habitados) en otras zonas de la galaxia y más allá. Así que imaginamos que la ruta hacia el oxígeno ha ocurrido en lugares distintos de la Tierra. Yo tiendo a creer que, contando con luz, agua, nutrientes y tiempo, la fotosíntesis oxigénica probablemente evolucionaría en otros mundos. Puede que la ruta hacia la producción de oxígeno fuera diferente, pero uno de los aspectos extraordinarios de la vida en la Tierra es que los microbios han evolucionado hasta llevar a cabo casi todos los tipos imaginables de metabolismo que pueden aportar energía para el crecimiento. Bajo este punto de vista, la fotosíntesis oxigénica no es más que una de esas múltiples posibilidades.
El registro en la Tierra indica, sin embargo, que no basta con la producción de oxígeno para que el oxígeno se acumule en la atmósfera. Hay poco consenso sobre el tiempo en que evolucionaron las cianobacterias, pero hay evidencias muy convincentes de que ocurrió antes de la gran oxidación (el GOE) de la atmósfera terrestre entre 2300 y 2400 millones de años atrás. De hecho, las evidencias indican que las cianobacterias existieron en una atmósfera mayormente anóxica durante cientos de millones de años —si no durante mil millones de años o más— antes de la oxigenación general de la atmósfera. Por lo que conocemos ahora, las mismas agitaciones de la Tierra que hacen de ella un planeta inmensamente habitable suministran también gases reductores (el más importante es el H2) a la superficie de la Tierra. Estos gases reaccionan con el oxígeno, y en la Tierra joven se eyectaban a unas tasas suficientes para eliminar todo el oxígeno liberado mediante el enterramiento del carbono orgánico y el azufre pirítico en los sedimentos. A medida que la Tierra se iba enfriando, la agitación tectónica del planeta se fue ralentizando, y de esto resultó una disminución del flujo de gases reductores desde el manto. Bajo este punto de vista, el GOE marca el punto en la historia de la Tierra en que el flujo de gases reductores (principalmente el H2) desde el manto bajó hasta una tasa menor que la tasa de liberación de oxígeno a la atmósfera. Sólo entonces pudo el oxígeno acumularse. También hay evidencias de que el GOE se prefiguró con vaivenes y ocasionales «tufillos» de oxígeno atmosférico, que empezaron 300 o 400 millones de años antes. Si ponemos números en esto, los niveles de base del oxígeno antes del GOE eran probablemente de una milésima del 1% de los actuales (un 0,001%) o menos, y durante los tufillos pudieron aumentar hasta alcanzar el 0,01% o 0,1% de los actuales (véase la figura 12.1 para una reconstrucción de la historia del oxígeno en la Tierra).
El GOE en sí mismo parece haber dado lugar a profundos cambios en el reciclado de nutrientes y carbono, con unos resultados sorprendentemente no lineales. Primero, parece que la movilización de nutrientes, posiblemente de fósforo, en una atmósfera recién oxigenada aceleró la producción de materia orgánica en los océanos, generando altas tasas de enterramiento de carbono orgánico y la mayor anomalía positiva de isótopos del carbono (la anomalía isotópica de Lomagundi) de toda la historia de la Tierra. También produjo una probable elevación de los niveles atmosféricos de oxígeno más allá de los producidos durante los estadios iniciales del GOE, quizá incluso aproximándose a los niveles actuales. Un enorme sumidero de oxígeno se generó cuando todo este carbono orgánico regresó, algo después, al entorno erosivo; esto disminuyó el nivel de oxígeno a unos valores muy bajos, aunque aparentemente algo mayores de los que eran típicos antes del GOE. Unos 500 millones de años después del GOE, el ciclo del carbono se estabilizó en un patrón relativamente estable, generando niveles de oxígeno que no excedían el 40% de los presentes, y que más probablemente estaban en el intervalo del 10% al 15% de los niveles actuales, o menos. De momento no se ha establecido con certeza un límite inferior durante este periodo, pero se sitúa probablemente entre el 0,1% y el 1% de los niveles actuales. Esto debería suponer el suficiente oxígeno para permitir la erosión química activa de la materia orgánica y el azufre pirítico en tierra firme, lo que en efecto parece haber sido el caso.
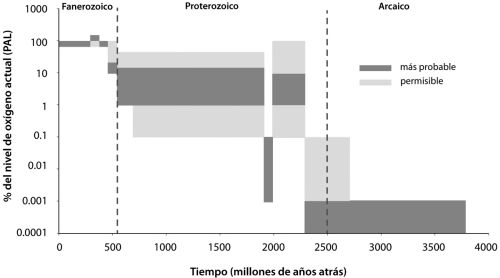
FIGURA 12.1 Sumario de la historia del oxígeno atmosférico a través del tiempo. Véase también Kump (2008).
La era neoproterozoica marca la transición desde la vida predominantemente microscópica de la Tierra temprana hacia la vida macroscópica del eón fanerozoico. La era neoproterozoica estuvo marcada por varias grandes glaciaciones, quizá globales, cuyo impacto en la historia del oxígeno en la Tierra es todavía incierto. Pese a ello, varios indicadores geoquímicos apuntan a una oxigenación de los océanos durante las fases finales de la era neoproterozoica. Esta oxigenación se correlaciona a trazos gruesos con la emergencia de los animales, incluidos los animales macroscópicos con motilidad. Tales organismos parecen requerir unos niveles de oxígeno del orden del 10% de los actuales (aunque muchos animales motiles modernos, especialmente los pequeños, pueden probablemente tolerar niveles tan bajos como el 1% de los actuales), y es tentador concluir que la emergencia de los animales fue posibilitada por un incremento de la oxigenación de los océanos. Si esto es cierto, hacia el final de la era neoproterozoica los niveles de oxígeno se elevaron desde unos valores cercanos al 1% de los actuales, o un poco mayores, hasta quizá el 10% o el 15% de los actuales.
Según otra hipótesis, los niveles de oxígeno estaban ya alrededor del 10% de los actuales antes de la emergencia de los animales. Bajo este punto de vista, los animales grandes evolucionaron en un entorno permisivo que ya existía previamente. De ser esto cierto, el retraso en la aparición de los animales sería consecuencia del hecho de que, simplemente, lleva tiempo para la evolución producir animales multicelulares complejos a partir de eucariotas unicelulares. De ser este el caso, la oxigenación de los océanos que revelan las mediciones geoquímicas no vino causada por un incremento del contenido de oxígeno en la atmósfera. En vez de eso, el registro de indicadores puede estar respondiendo a un incremento de la oxigenación de los océanos operado, posiblemente, por los propios animales. Este incremento en la oxigenación de los océanos puede verse como una extensión de las áreas oceánicas en que el oxígeno perdura. A mí me gusta bastante este punto de vista, ya que no hay rasgos obvios del ciclo del carbono que apunten a un incremento sostenido en la libaración de oxígeno a la atmósfera durante la era neoproterozoica. Futuras investigaciones resolverán esta disyuntiva.
Considero probable que los niveles de oxígeno durante las fases tempranas del eón fanerozoico estuvieran en el intervalo del 10% al 20% de los actuales. Esto es coherente con algunos modelos de evolución del oxígeno durante el eón fanerozoico. Las mediciones geoquímicas insinúan un incremento sustancial de la oxigenación del océano durante el silúrico tardío y el devónico temprano (hace unos 420 millones de años), coincidiendo con un drástico incremento en el tamaño de los peces en los océanos. Una explicación para esta correlación es que las mediciones geoquímicas hayan capturado un verdadero incremento en los niveles del oxígeno atmosférico, y que la talla extra grande de los peces fuera una consecuencia de los altos contenidos de oxígeno. Un verdadero incremento de los niveles de oxígeno atmosférico tiene bastante sentido, porque más o menos por ese tiempo surgieron y se extendieron las plantas terrestres. Los bajos requerimientos de nutrientes de las plantas terrestres, combinados con su resistente materia orgánica, habrían conducido a un enterramiento incrementado de materia orgánica y a una mayor liberación de oxígeno a la atmósfera. Según esta idea, la evolución de las plantas terrestres condujo a una reorganización fundamental del ciclo del carbono, lo que produjo unos niveles mucho más altos de oxígeno atmosférico, lo que a su vez tuvo efectos en cascada sobre la evolución biológica. Debido a las peculiaridades de la paleogeografía, se dieron unas tasas altísimas de enterramiento de materia orgánica en los masivos cenagales del carbonífero y principios del pérmico. Esta situación condujo a unas tasas altísimas de liberación de oxígeno, y durante esta fase de la historia de la Tierra las concentraciones de oxígeno atmosférico pueden muy bien haber superado significativamente los niveles actuales. El gigantismo de los insectos fue una consecuencia visible de estos altos niveles de oxígeno.
En cuanto al mundo moderno, nosotros estamos cómodamente adaptados a los niveles existentes de oxígeno atmosférico. Estos niveles son estables y, pese al uso de combustibles fósiles, los cambios en la concentración de oxígeno atmosférico, aunque detectables, resultarán triviales hasta que unos procesos geológicos/biológicos combinados y de escala global conspiren para cambiarlos. Esto probablemente llevaría millones de años. Así que respira profundamente y reflexiona. Hay una historia tremenda detrás de los niveles de oxígeno que disfrutamos en la atmósfera de la Tierra.
Bibliografía
PREFACIO - Gradstein, F., J. Ogg, A. Smith, 2004. A Geologic Time Scale. Cambridge University Press, Cambridge, Reino Unido.
- Hutton, J., 1788. Theory of the Earth. Royal Society of Edinburgh, Edimburgo. (Libro clásico que, según muchos, marcó el comienzo de la geología moderna).
CAPÍTULO 1- Baross, J. A., S. A. Benner, G. D. Cody, S. D. Copley, N. R. Pace, J. H. Scott, R. Shapiro, M. L. Sogin, J. L. Stein, R. E. Summons, J. W. Szostak, 2007. «The Limits of Organic Life in Planetary Systems». National Academy of Sciences, Washington. (La Academia Nacional de Ciencias de Estados Unidos informa sobre los límites de la vida. Véase el capítulo 6 sobre la importancia del agua, con especulaciones sobre otros posibles solventes para la vida).
- Canfield, D. E., E. Kristensen, B. Thamdrup, 2005. Aquatic Geomicrobiology. Academic Press, Ámsterdam. (Véase la discusión sobre las reacciones redox y la vida en el capítulo 3.)
- Harland, D. M., 2005. Water and the Search for Life on Mars. Springer, Berlín. (Un libro popular que resume las evidencias de agua en Marte e incluye los primeros resultados de la misión Mars Exploration Rover, MER).
- Kasting, J. F., 1993. «Habitable zones around main sequence stars». ICARUS 101, pp. 108-128. (Excelente discusión de la zona habitable, incluyendo una buena perspectiva histórica).
- Kasting, J. F., 2010. How to Find a Habitable Planet. Princeton University Press, Princeton, NJ. (Explicación legible y entretenida de la evolución de la atmósfera y el clima de la Tierra, y de la búsqueda de mundos habitables en otros lugares del sistema solar).
- Knoll, A. H., 2003. Life on a Young Planet. The First Three Billion Years of Evolution on Earth. Princeton University Press, Princeton, NJ, y Oxford, Reino Unido. (Un viaje fantástico por los primeros 3000 millones de años de vida en la Tierra).
- Mustard, J. F., S. L. Murchie, S. M. Pelkey, B. L. Ehlmann, R. E. Milliken y col., 2008. «Hydrated silicate minerals on mars observed by the Mars reconnaissance orbiter CRISM instrument». Nature 454, pp. 305-309. (Evidencias espectroscópicas de minerales de silicato hidratado que implican erosión en un fluido acuoso).
- Rosing, M. T., D. K. Bird, N. H. Sleep, C. J. Bjerrum, 2010. «No climate paradox under the faint early Sun». Nature 464, pp. 744-747. (Rosing y sus colegas argumentan que, debido al bajo albedo de la Tierra joven, no hay de hecho ninguna paradoja del joven Sol débil).
- Sagan, C., G. Mullen, 1972. «Earth and Mars: Evolution of atmospheres and surface temperatures». Science 177, pp. 52-56. (Sagan y Mullen presentan la paradoja del joven Sol débil).
- Squyres, S. W., R. E. Arvidson, J. F. Bell, F. Calef, B. C. Clark y col., 2012. «Ancient impact and aqueous processes at Endeavour Crater, Mars». Science 336, pp. 570-576. (Evidencias recientes de agua obtenidas por la misión MER).
- Walker, J.C.G., P. B. Hays, J. F. Kasting, 1981. «A negative feedback mechanism for the long-term stabilization of Earth’s surface temperature». Journal of Geophysical Research-Oceans and Atmospheres 86, pp. 9776-9782. (Se introduce una retroalimentación negativa a través del ciclo del carbono para resolver la paradoja del joven Sol débil).
CAPÍTULO 2- Baas Becking, L.G.M., 1925. «Studies on the sulphur bacteria». Annals of Botany 39, pp. 613-650. (Presenta por primera vez la idea del sulfureto). —, 1934. Geobiologie: of Inleiding tot de Milieukunde. W.P. Van Stockum & Zoon N. V., Den Haag, Holanda. (Libro clásico que define la geobiología como campo de estudio).
- Brock, T. D., 1994. Life at High Temperatures. Yellowstone Association for Natural Science, History & Education, Yellowstone National Park, WY. (Introducción popular a la vida en las fumarolas hidrotermales, centrada en el parque de Yellowstone, escrita por Thomas Brock, el pionero en el campo).
- Canfield, D. E., M. T. Rosing, C. Bjerrum, 2006. «Early anaerobic metabolisms». Philosophical Transactions of the Royal Society B 361, pp. 1819-1834. (Un intento de restringir el nivel de actividad de la biosfera antes de la fotosíntesis oxigénica).
- Crowe, S. A., C. Jones, S. Katsev, C. Magen, A. H. O’Neill, A. Sturm, D. E. Canfield y col., 2008. «Photoferrotrophs thrive in an Archean Ocean analogue». Proceedings of the National Academy of Sciences of the United States of America 105, pp. 15938-15943. (Un posible análogo del océano primitivo con aguas ricas en hierro que probablemente sostenían fotótrofos anoxigénicos oxidantes de hierro).
- David, L. A., E. J. Alm, 2011. «Rapid evolutionary innovation during an Archaean genetic expansion». Nature 469, pp. 93-96. (Impresionante mirada a la evolución de las rutas metabólicas).
- Kharecha, P., J. Kasting, J. Siefert, 2005. «A coupled atmosphere-ecosystem model of the early Archean Earth». Geobiology 3, pp. 53-76. (Una inteligente utilización de los modelos del oxígeno atmosférico para explorar la actividad de la biosfera de la Tierra joven).
- Shen, Y., R. Buick, D. E. Canfield, 2001. «Isotopic evidence for microbial sulphate reduction in the early Archean era». Nature 410, pp. 77-81. (Las evidencias más tempranas de reducción microbiana del sulfato).
- Shen, Y. N., J. Farquhar, A. Masterson, A. J. Kaufman, R. Buick, 2009. «Evaluating the role of microbial sulfate reduction in the early Archean using quadruple isotope systematics». Earth and Planetary Science Letters 279, pp. 383-391. (Evidencias adicionales de la evolución primitive de la reducción microbiana de sulfato).
- Ueno, Y., K. Yamada, N. Yoshida, S. Maruyama, Y. Isozaki, 2006. «Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era». Nature 440, pp. 516-519. (Evidencias de la evolución temprana de la metanogénesis).
- Widdel, F., S. Schnell, S. Heising, A. Ehrenreich, B. Assmus, B. Schink, 1993. «Ferrous iron oxidation by anoxygenic phototrophic bacteria». Nature 362, pp. 834-835. (Primera descripción de la oxidación fototrófica anoxigénica del Fe2+.)
CAPÍTULO 3- Allen, J. F., 2005. «A redox switch for the origin of two light reactions in photosynthesis». FEBS Letters 579, pp. 963-968. (Allen presenta aquí su hipótesis sobre el origen de los centros de reacción acoplados en los fotótrofos oxigénicos).
- Allen, J. F., W. Martin, 2007. «Out of thin air». Nature 445, pp. 610-612. (Buen resumen de la evolución de la fotosíntesis oxigénica, con un interesante rompecabezas sobre el desarrollo del racimo de manganeso).
- Badger, M. R., E. J. Bek, 2008. «Multiple Rubisco forms in proteobacteria: Their functional significance in relation to CO2 acquisition by the CBB cycle». Journal of Experimental Botany 59, pp. 1525-1541. (Excelente revision de Rubisco, sus múltiples formas y su actividad catalítica).
- Blankenship, R. E., 1992. «Origin and early evolution of photosynthesis». Photosynthesis Research 33, pp. 91-111. (Los artículos originales que describieron la evolución de PSI y PSII a partir de los centros de reacción que usan los fotótrofos anoxigénicos).
- —, 2010. «Early evolution of photosynthesis». Plant Physiology 154, pp. 434-438. (Excelente revisión de la evolución de la fotosíntesis). —, H. Hartman, 1998. «The origin and evolution of oxygenic photosynthesis». Trends in Biochemical Sciences 23, pp. 94-97. (Describe la evolución de la fotosíntesis oxigénica y del complejo de evolución del oxígeno a partir de las enzimas catalasas).
- Canfield, D. E., E. Kristensen, B. Thamdrup, 2005. Aquatic Geomicrobiology. Academic Press, Ámsterdam. (Consúltese el capítulo 4 para una revision de la fotosíntesis).
- Falkowski, P. G., J. A. Raven, 2007. Aquatic Photosynthesis. Princeton Unversity Press, Princeton, NJ. (El libro que cubre todos los aspectos de la fotosíntesis acuática).
- Hohmann-Marriott, M. F., R. E. Blankenship, 2011. «Evolution of photosynthesis», Annual Review of Plant Biology 62, 515-548. (Revisión reciente de la evolución de la fotosíntesis).
- Raymond, J., Blankenship, R. E., 2008. «The origin of the oxygen-evolving complex». Coordination Chemical Reviews 252, 377-383. (Estudio estructural de la similitud entre el complejo de evolución del oxígeno y la catalasa Mn).
- Sadekar, S., J. Raymond, R. E. Blankenship, 2006. «Conservation of distantly related membrane proteins: Photosynthetic reaction centers share a common structural core». Molecular Biology and Evolution 23, 2001-2007. (Estudio que compara todas las estructuras de los centros de reacción fotosintéticos).
- Tabita, F. R., T. E. Hanson, H. Y. Li, S. Satagopan, J. Singh, S. Chan, 2007. «Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs». Microbiology and Molecular Biology Reviews 71, 576-599. (Excelente revisión de las proteínas Rubisco y similares a Rubisco).
- Xiong, J., W. M. Fischer, K. Inoue, M. Nakahara, C. E. Bauer, 2000. «Molecular evidence for the early evolution of photosynthesis». Science 289, 1724-1730. (Estudio molecular sobre la evolución de las rutas de formación de los pigmentos fotosintéticos).
CAPÍTULO 4- Berman-Frank, I., P. Lundgren, Y.-B. Chen, H. Küpper, Z. Kolber, B. Bergman, P. Falkowski, 2001. «Segregation of nitrogen fixation and oxygenic photosynthesis in the marine cyanobacterium Trichodesmium». Nature 294, 1534-1537. (Brillante trabajo sobre la regulación de la fijación de nitrógeno por el más importante fijador de nitrógeno de los océanos).
- Budel, B., B. Weber, M. Kuhl, H. Pfanz, D. Sultemeyer, D. Wessels, 2004. «Reshaping of sandstone surfaces by cryptoendolithic cyanobacteria: Bioalkalization causes chemical weathering in arid landscapes». Geobiology 2, 261-268. (Buena revisión sobre las cianobacterias endolíticas, incluyendo dónde viven y su influencia en las rocas).
- Canfield, D. E., D. J. Des Marais, 1991. «Aerobic sulfate reduction in microbial mats». Science 251, 1471-1473. (Descripción de la reducción de sulfato bajo condiciones óxicas en una alfombra microbiana, con una descripción de la estructura de la alfombra microbiana). —, D. J. Des Marais, 1993. «Biogeochemical cycles of carbon, sulfur, and free oxygen in a microbial mat». Geochimica et Cosmochimica Acta 57, 3971-3984. (Descripción de los ciclos del carbono y el sulfuro en un ecosistema de alfombra microbiana). —, E. Kristensen, B. Thamdrup, 2005. Aquatic Geomicrobiology. Academic Press. Ámsterdam. (Véase el capítulo 7 para una revisión del ciclo del nitrógeno, y el capítulo 13 para una revisión de las alfombras microbianas de cianobacterias).
- Chisholm, S. W., R. J. Olson, E. R. Zettler, R. Goericke, J. B. Waterbury, N. A. Welschmeyer, 1988. «A novel free-living prochlorophyte abundant in the oceanic euphotic zone». Nature 334, 340-343. (Primera descripción de Prochlorococcus en los océanos).
- Curtis, S. E., M. T. Clegg, 1984. «Molecular evolution of chloroplast DNS-sequences». Molecular Biology and Evolution 1, 291-301. (Evidencia molecular inicial que asocia los cloroplastos a las cianobacterias).
- Des Marais, D. J., 1990. «Microbial mats and the early evolution of life». Tree 5, 140-144. (Buena revisión inicial de las alfombras microbianas y su relevancia para el estudio de los entornos de la Tierra primitiva).
- Garcia-Pichel, F., J. Belnap, S. Neuer, F. Schanz, 2003. «Estimates of global cyanobacterial biomass and its distribution». Algological Studies 109, 213-227. (Revisión cuantitativa de la importancia de las cianobacterias en diversos ecosistemas y a escala global).
- Johnson, P. W., J. M. Sieburth, 1979. «Chroococcoid cyanobacteria in the sea: Ubiquitous and diverse phototropic biomass». Limnology and Oceanography 24, 928-935. (Primera descripción de las cianobacterias marinas que resultaron ser Prochlorococcus).
- Johnson, Z. I., E. R. Zinser, A. Coe, N. P. McNulty, E. M. S. Woodward, S. W. Chisholm, 2006. «Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients». Science 311, 1737-1740. (Maravillosa descripción de la distribución de varias cepas de Prochlorococcus según la profundidad en el océano, y también en función de la latitud).
- Jørgensen, B. B., D. J. Des Marais, 1988. «Optical properties of benthic photosynthetic communities: Fiber-optic studies of cyanobacterial mats». Limnology and Oceanography 33, 99-113. (Estudio inicial de las propiedades ópticas de las alfombras microbianas, posibilitado por el desarrollo de microsensores especiales de fibra óptica).
- Kettler, G. C., A. C. Martiny, K. Huang, J. Zucker, M. L. Coleman, S. Rodrigue, F. Chen, F. y col., 2007. «Patterns and implications of gene gain and loss in the evolution of Prochlorococcus». PLOS Genetics 3, 2515-2528. (Buena discusión de la plasticidad del genoma de Prochlorococcus).
- Khakhina, L. N., 1992. Concepts of Symbiogenesis: Historical and Critical Study of the Research of Russian Scientists. Yale University Press, New Haven, CT. (Resumen de las contribuciones de Konstantin Sergeevich Merezhkovsky y otros científicos rusos al desarrollo de la teoría endosimbiótica del desarrollo de la célula eucariota).
- Liu, H., H. A. Nolla, L. Campbell, 1997. «Prochlorococcus growth rate and contribution to primary production in the equatorial and subtropical North Pacific Ocean». Aquatic Microbial Ecology 12, 39-47. (Estimaciones de la contribución de Prochlorococcus a la producción primaria marina).
- Revsbech, N. P., 1989. «An Oxygen Microsensor with a guard cathode». Limnology and Oceanography 34, 474-478. (Descripción del microelectrodo de oxígeno más utilizado en la investigación ecológica). —, B. B. Jørgensen, T. H. Blackburn, Y. Cohen, 1983. «Microelectrode studies of the photosynthesis and O2, H2S, and pH profiles of a microbial mat». Limnology and Oceanography 28, 1062-1074. (Brillante estudio de la dinámica del oxígeno y el azufre en las alfombras microbianas usando los microelectrodos recién desarrollados).
- Sagan, L., 1967. «On the origin of mitosing cells». Journal of Theoretical Biology 14, 225-274. (Primera exposición por Lynn Margulis, entonces Lynn Sagan, de la teoría endosimbiótica del origen de cloroplastos y mitocondrias).
- Severin, I., L. J. Stal, 2010. «Spatial and temporal variability in nitrogenase activity and diazotrophic community composition in coastal microbial mats». Marine Ecology-Progress Series 417, 13-25. (Estudio que muestra las varias estrategias diferentes que utilizan las comunidades bénticas fijadoras de nitrógeno).
- Sohm, J. A., E. A. Webb, D. G. Capone, 2011. «Emerging patterns of marine nitrogen fixation». Nature Reviews Microbiology 9, 499-508. (Buena revision de la fijación de nitrógeno en los océanos).
- Waterbury, J. B., S. W. Watson, R. R. L. Guillard, L. E. Brand, 1979. «Widespread occurrence of a unicellular, marine, planktonic, cyanobacterium». Nature 277, 293-294. (Primera descripción de Synecococcus en los océanos).
- Zobell, C. E., 1946. Marine Microbiology. Chronica Botanica Company, Waltham, MA. (Libro clásico sobre la microbiología de los mares).
CAPÍTULO 5- Berner, R. A., 1987. «Models for carbon and sulfur cycles and atmospheric oxygen: Application to Paleozoic geologic history». American Journal of Science 287, 177-196. (Primer artículo de Bob sobre la regulación del oxígeno, en el que introdujo el concepto de reciclado rápido). —, 2004. The Phanerozoic Carbon Cycle: CO2 and O2. Oxford University Press, Oxford, Reino Unido. (Tratamiento completo del modelado a largo plazo del CO2 y el O2 en la atmósfera, basado en gran medida en el propio trabajo de Bob Berner). —, K. A. Maasch, 1996. «Chemical weathering and controls on atmospheric O2 and CO2: Fundamental principles were enunciated by J. J. Ebelman in 1845». Geochimica et Cosmochimica Acta 60, 1633-1637. (Relato histórico del papel de Ebelman en el esclarecimiento de los controles de la concentración de oxígeno atmosférico).
- Canfield, D. E., 2005. «The early history of atmospheric oxygen: Homage to Robert M. Garrels». Annual Review of Earth and Planetary Science 33, 1-36. (Una vision general de la historia del oxígeno atmosférico, con un punto de entrada en el trabajo de Bob Garrels).
- Ebelman, J. J., 1845. «Sur les produits de la décomposition des especes minérales de la famille des silicates». Annales des Mines 7, 3-66. (Primera exposición de los controles sobre la concentración de oxígeno atmosférico).
- Fennel, K., Follows, M., Falkowski, P. G., 2005. «The co-evolution of the nitrogen, carbon and oxygen cycles in the Proterozoic ocean». American Journal of Science 305, 526-545. (Un estudio de modelado sobre el papel del nitrógeno en el control de la producción marina primaria durante los periodos de la historia de la Tierra en que las profundidades oceánicas eran mayormente anóxicas).
- Garrels, R. M., E. A. Perry, 1974. «Cycling of carbon, sulfur, and oxygen through geologic time», en: Goldberg, E. D. (editor), The Sea. John Wiley and Sons, Nueva York, pp. 303-336. (Clásica exposición moderna del reciclado de elementos y de los procesos que controlan la concentración de oxígeno).
- Holland, H. D., 1978. The Chemistry of the Atmosphere and Oceans. John Wiley and Sons, Nueva York. (Texto clásico sobre los procesos que operan para controlar la química de la atmósfera y los océanos).
- Ingall, E., R. Jahnke, 1994. «Evidence for enhanced phosphorus regeneration from marine sediments overlain by oxygen depleted waters». Geochimica et Cosmochimica Acta 58, 2571-2575. (Presenta evidencias que apoyan la idea original de Ingall de que el fósforo se regenera preferentemente a partir de los sedimentos que subyacen a una columna de agua oxigenada).
- Kump, L. R., 1988. «Terrestial feedback in atmospheric oxygen regulation by fire and phosphorus». Nature 335, 152-154. (El papel del fuego en la regulación del oxígeno atmosférico es tomado en serio).
- Murray, J. W., L. A. Codispoti, G. E. Friederich, 1995. «Oxidation-reduction environments: The suboxic zone in the Black Sea». Advances in Chemistry Series 244, 157-176. (Buena revision de la química de la columna de agua del mar Negro).
- Petsch, S. T., R. A. Berner, T. I. Eglinton, 2000. «A field study of the chemical weathering of ancient sedimentary organic matter». Organic Geochemistry 31, 475-487. (Precioso estudio de campo de la erosión oxidativa del carbono orgánico y el azufre durante la formación del suelo).
- Richards, F. A., 1965. «Anoxic basins and fjords», en: Riley, J. P., G. Skirrow, (editores), Chemical Oceanography. Academic Press, Londres, pp. 611-645. (Exposición clásica de la química de las cuencas y fiordos anóxicos).
- Van Cappellen, P., E. D. Ingall, 1996. «Redox stabilization of the atmosphere and oceans by phosphorus-limited marine productivity». Science 271, 493-496. (Estudio de modelado que muestra cómo la retroalimentación por fósforo de Ingall puede funcionar para regular los niveles de oxígeno).
- Watson, A., J. E. Lovelock, L. Margulis, 1978. «Methanogenesis, fires and the regulation of atmospheric oxygen». Biosystems 10, 293-298. (Primer estudio de la influencia de las concentraciones de oxígeno en los incendios).
CAPÍTULO 6- Brasier, M. D., O. R. Green, A. P. Jephcoat, A. K. Kleppe, M. J. van Krankendonk, J. F. Lindsay, A. Steele, N. V. Grassineau, 2002. «Questioning the evidence for Earth’s oldest fossils». Nature 416, 76-81. (Este artículo representa el primer asalto de Martin Brasier sobre el carácter biogénico de los microfósiles de 3450 millones de años de Bill Schopf).—, O. R. Green, J. F. Lindsay, N. McLoughlin, A. Steele, C. Stoakes, 2005. «Critical testing of earth’s oldest putative fossil assemblage from the similar to 3.5 Ga Apex Chert, Chinaman Creek, Western Australia». Precambrian Research 140, 55-102. (Este artículo es una respuesta a la réplica de Schopf a la crítica original de Brasier sobre los fósiles de Schopf. Brasier usa aquí, entre otras cosas, espectroscopía Raman para argumentar que los «fósiles» del Apex Chert tienen un origen no biológico).
- Rosing, M. T., 1999. «13C-depleted carbon microparticles in >3700-Ma sea-floor sedimentary rocks from West Greenland». Science 283, 674-676. (Minik Rosing presenta evidencias con isótopos de carbono de vida en las rocas sedimentarias primitivas de Isua, Groenlandia).
- Schopf, J. W., 1993. «Microfossils of the early Archean Apex Chert: New evidence of the antiquity of life». Science 260, 640-646. (Bill Schopf presenta sus imágenes icónicas de lo que interpretó como posibles microfósiles cianobacterianos del Apex Chert de 3450 millones de años, en Australia occidental).
- A. B. Kudryavtsev, 2009. «Confocal laser scanning microscopy and Raman imagery of ancient microscopic fossils». Precambrian Research 173, 39-49. (Schopf presenta más evidencias con láser Raman de que las estructuras del Apex Chert son antiguos fósiles microbianos).—, A. B. Kudryavtsev, 2012. «Biogenicity of Earth’s earliest fossils: A resolution of the controversy». Gondwana Research 22, 761-771. (Schopf y Kudryavtsev aportan evidncias adicionales del carácter biogénico de los fósiles del Apex Chert). —, A. B. Kudryavtsev, D. G. Agresti, T. J. Wdowiak, A. D. Czaja, 2002. «Laser-Raman imagery of Earth’s earliest fossils». Nature 416, 73-76. (Schopf utiliza espectroscopía Raman para refutar los argumentos de Brasier de que las estructuras que Schopf describió originalmente en el Apex Chert no son biogénicas).
- Summons, R. E., A. S. Bradley, L. L. Jahnke, J. R. Waldbauer, 2006. «Steroids, triterpenoids and molecular oxygen». Philosophical Transactions of the Royal Society B 361, 951-968. (Una gran revisión del papel del oxígeno en la síntesis de esteroles).
- Van Kranendonk, M. J., 2006. «Volcanic degassing, hydrothermal circulation and the flourishing of early life on Earth: A review of the evidence from c. 3490-3240 Ma rocks of the Pilbara Supergroup, Pilbara Craton, Western Australia». Earth-Science Reviews 74, 197-240. (Incluye una revisión de las evidencias del origen hidrotermal de las rocas que contienen a los fósiles del Apex Chert).
- Vernadsky, V. I., 1998. The Biosphere. Springer-Verlag New York, Nueva York. (Libro clásico que argumenta a favor del papel de la vida en dar forma a la geosfera y la atmósfera).
- Waldbauer, J. R., D. K. Newman, R. E. Summons, 2011. «Microaerobic steroid biosynthesis and the molecular fossil record of Archean life». Proceedings of the National Academy of Sciences of the United States of America 108, 13409-13414. (Un impresionante estudio que demuestra que sólo se necesitan trazas de oxígeno para la síntesis de esteroides por los organismos aeróbicos). —, L. S. Sherman, D. Y. Sumner, R.E. Summons, 2009. «Late Archean molecular fossils from the Transvaal Supergroup record the antiquity of microbial diversity and aerobiosis». Precambrian Research 169, 28-47. (Precioso estudio con biomarcadores, incluídos los esteranos, en rocas de 2460 a 2670 millones de años, y su relación con el oxígeno).
CAPÍTULO 7- Anbar, A. D., Y. Duan, T. W. Lyons, G. L. Arnold, B. Kendall, R. A. Creaser, A. J. Kaufman y col., 2007. «A whiff of oxygen before the Great Oxidation Event?». Science 317, 1903-1906. (Una bella mirada a la geoquímica de las pizarras de 2500 millones de años del monte McRae, que muestra las evidencias de una leve oxidación de la atmósfera de la Tierra antes del GOE. Aquí se acuña el término «whiff», o tufillo).
- Farquhar, J., H. M. Bao, M. Thiemens, 2000. «Atmospheric influence of Earth’s earliest sulfur cycle». Science 289, 756-758. (Artículo definitivo que muestra la ocurrencia común de una señal isotópica de azufre independiente de masa antes del GOE, y su desaparición después de él. La mejor evidencia disponible de un bajo nivel de oxígeno atmosférico antes del GOE). —, J. Savarino, S. Airieau, M. H. Thiemens, 2001. «Observation of the wavelength-sensitive mass-dependent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere». Journal of Geophysical Research 106, 32829-32839. (Sagaces experimentos fotoquímicos que asocian la señal isotópica de azufre independiente de masa con las reacciones fotoquímicas con radiación ultravioleta).
- Frimmel, H. E., 2005. «Archaean atmospheric evolution: Evidence from the Witwatersrand gold fields, South Africa». Earth-Science Reviews 70, 1-46. (Revisión moderna de la uraninita y la pirita de depósitos aluviales en las minas de Witwatersrand, y su relevancia para la evolución atmosférica).
- Holland, H. D., 1962. «Model for the evolution of the Earth’s atmosphere», en: Engel, A. E. J., H. L. James, B. F. Leonard (editores), Petrologic Studies: A Volume in Honor of A. F. Buddington. Geological Society of America, Boulder, CO, pp. 447-477. (Asombroso artículo que expone el pensamiento temprano de Dick Holland sobre la evolución de las concentraciones atmosféricas de oxígeno. Este artículo sentó las bases de gran parte de su trabajo posterior). —, 1984. The Chemical Evolution of the Atmosphere and Oceans. Princeton University Press, Princeton, NJ. (Libro clásico que describe los puntos de vista de Dick sobre la evolución de la química atmosférica y oceánica hacia 1984. Todavía utilizado ampliamente). —, 2009. «Why the atmosphere became oxygenated: A proposal». Geochimica et Cosmochimica Acta 73, 5241-5255. (Brillante artículo que ofrece una explicación plausible del GOE).
- Pavlov, A. A., J. F. Kasting, 2002. «Mass-independent fractionation of sulfur isotopes in Archean sediments: Strong evidence for an anoxic Archean atmosphere». Astrobiology 2, 27-41. (Modelo de la química atmosférica que calibra los niveles de oxígeno necesarios para crear fraccionamientos de isótopos de azufre independientes de masa).
- Phillips, G. N., J. D. M. Law, R. E. Myers, 2001. «Is the redox state of the Archean atmosphere constrained?». SEG Newsletter 47, 8-18. (Un punto de vista escéptico sobre el origen en detritos de las uraninitas y piritas de Witwatersrand).
- Rasmussen, B., R. Buick, 1999. «Redox state of the Archean atmosphere: Evidence from detrital heavy minerals in ca. 3250-2750 Ma sandstones from the Pilbara Craton, Australia». Geology 27, 115-118. (Presenta más casos de pirita y uraninita de detritos durante el eón arcaico).
- Utter, T., 1980, «Rounding of ore particles from the Witwatersrand gold and uranium deposit (South Africa) as an indicator of their detrital origin». Journal of Sedimentary Petrology 71-76. (Buen estudio petrográfico y de microscopía electrónica de barrido sobre los granos de uraninita y pirita de detritos del Witwatersrand).
- Wille, M., J. D. Kramers, T. F. Nagler, N. J. Beukes, S. Schroder, T. Meisel, J. P. Lacassie, A. R. Voegelin, 2007. «Evidence for a gradual rise of oxygen between 2.6 and 2.5 Ga from Mo isotopes and Re-PGE signatures in shales». Geochimica et Cosmochimica Acta 71, 2417-2435. (Presenta la primera evidencia geoquímica de un «tufillo» de oxígeno antes del GOE).
CAPÍTULO 8- Catling, D. C., M. W. Claire, 2005, «How the Earth’s atmosphere evolved to an oxic state: A status report». Earth and Planetary Science Letters 237, 1-20. (Un buen resumen de los controles de la regulación del oxígeno atmosférico, y de las explicaciones del GOE, como se entendían en 2005). —, K. J. Zahnle, C. P. McKay, 2001. «Biogenic methane, hydrogen escape, and the irreversible oxidation of early life». Science 293, 839-843. (Un modelo muy inteligente con el que Catling y sus colegas explican el GOE mediante el efecto acumulado de la fotolisis del metano para dar H2, y la pérdida de H2 al espacio, conduciendo a la oxidación del entorno de superficie).
- Cloud, P. E., 1968. «Atmospheric and hydrospheric evolution on the primitive earth». Science 160, 729-736. (Artículo «pensando a lo grande» que describe los principales estadios de la evolución química y biológica de la Tierra). —, 1972. «A working model of the primitive Earth». American Journal of Science 272, 537-548. (Desarrolla las ideas presentadas en el artículo de 1968).
- Crowell, J. C., 1995. «Preston Cloud, 1912-1991: A biographical memoir», en: Biographical Memoirs. National Academy of Sciences Press, Washington, DC, pp. 43-63. (Preciosa biografía de Preston Cloud).
- Guo, Q. J., H. Strauss, A. J. Kaufman, S. Schroder, J. Gutzmer, B. Wing, M. A. Baker y col., 2009. «Reconstructing Earth’s surface oxidation across the Archean-Proterozoic transition». Geology 37, 399-402. (Refina la datación del GOE mediante evidencias con isótopos de azufre en rocas sedimentarias de la edad huroniana).
- Holland, H. D., 1994. «Early Proterozoic atmospheric change», en: Bengston, S. (editor), Early Life on Earth. Columbia University Press, Nueva York, pp. 237-244. (Buen resumen de las evidencias del GOE). —, 2009. «Why the atmosphere became oxygenated: A proposal». Geochimica et Cosmochimica Acta 73, 5241-5255. (La propuesta de «valoración» de Holland para el GOE, donde el O2 se eleva cuando el flujo de gases reducidos del manto cae por debajo de la tasa de liberación de O2 por enterramiento de carbono orgánico y pirita).
- Karhu, J. A., H. D. Holland, 1996. «Carbon isotopes and the rise of atmospheric oxygen». Geology 24, 867-870. (Una compilación de las evidencias con isótopos de carbono que demarcan la anomalía isotópica de Lomagundi, a la que originalmente se consideró la causa del GOE).
- Kasting, J. F., D. H. Eggler, S. P. Raeburn, 1993. «Mantle redox evolution and the oxidation state of the archean atmosphere». Journal of Geology 101, 245-257. (En una inteligente propuesta, Kasting y sus colegas aducen que el manto era más reductor en la Tierra joven, permitiendo una atmósfera reductora incluso a pesar de la producción de oxígeno por las cianobacterias).
- Kirschvink, J. L., R. E. Kopp, 2008. «Palaeoproterozoic ice houses and the evolution of oxygen-editing enzymes: The case for a late origin of photosystem II». Philosophical Transactions of the Royal Society B 363, 2755-2765. (Kirschvink y Kopp argumentan aquí que el GOE fue el resultado de la evolución de las cianobacterias hace unos 2300 millones de años, y discuten y refutan las evidencias de que las cianobacterias son anteriores a esa fecha).
- Kump, L. R., J. F. Kasting, M. E. Barley, 2001. «Rise of atmospheric oxygen and the “upside-down” archean mantle». Geochemistry Geophysics Geosystems vol. n.º 2, paper number 2000GC000114. (Kump y sus colegas asocian el GOE al cambio del manto, donde los gases volcánicos se originan a partir de fuentes regionales más oxidadas que antes, cuando la atmósfera era reductora).
- Sekine, Y., K. Suzuki, R. Senda, K. T. Goto, E. Tajika, R. Tatad, K. Goto y col., 2011. «Osmium evidence for synchronicity between a rise in atmospheric oxygen and Paleoproterozoic deglaciation». Nature Communications vol. n.º 2. doi:10.1038/ncomms1507. (Buen estudio que aporta evidencias de la movilidad del osmio como resultado del GOE).
CAPÍTULO 9- Arnold, G. L., A. D. Anbar, J. Barling, T. W. Lyons, 2004. «Molybdenum isotope evidence for widespread anoxia in mid-Proterozoic oceans». Science 304, 87-90. (Primer artículo que demuestra que los isótopos de Mo pueden restringir la extensión de las condiciones euxínicas en los océanos primitivos).
- Bekker, A., J. F. Slack, N. Planavsky, B. Krapez, A. Hofmann, K. O. Konhauser, O. J. Rouxel, 2010. «Iron formation: The sedimentary product of a complex interplay among mantle, tectonic, oceanic, and biospheric processes». Economic Geology 105, 467-508. (Una buena revisión contemporánea de las formaciones de hierro bandeado).
- Cameron, E. M., 1982. «Sulphate and sulphate reduction in early Precambrian oceans». Nature 296, 145-148. (Encuentra altos fraccionamientos de isótopos de azufre después del GOE, y postula un aumento en las concentraciones marinas de sulfato).
- Canfield, D. E., 1998. «A new model for Proterozoic ocean chemistry». Nature 396, 450-453. (Propone que la oxigenación de la atmósfera hizo que los océanos se volvieran más sulfurosos, causando una remoción masiva del hierro disuelto y poniendo fin a la deposición de formaciones de hierro bandeado). —, 2005. «The early history of atmospheric oxygen: Homage to Robert M. Garrels». Annual Review of Earth and Planetary Science 33, 1-36. (Revisa la historia del oxígeno atmosférico y los procesos que regulan su concentración). —, S. W. Poulton, A. H. Knoll, G. M. Narbonne, G. Ross, T. Goldberg, H. Strauss, 2008. «Ferruginous conditions dominated later Neoproterozoic deep-water chemistry». Science 321, 949-952. (Documenta las generalizadas condiciones ferruginosas de las aguas oceánicas profundas en la era neoproterozoica). —, A. Teske, 1996. «Late Proterozoic rise in atmospheric oxygen concentration inferred from phylogenetic and sulphur-isotope studies». Nature 382, 127-132. (Evidencias con isótopos de azufre de un aumento neoproterozoico en las concentraciones de oxígeno atmosférico).
- Frei, R., C. Gaucher, S. W. Poulton, D. E. Canfield, 2009. «Fluctuations in Precambrian atmospheric oxygenation recorded by chromium isotopes». Nature 461, 250-253. (Utiliza isótopos de Cr para documentar fluctuaciones en la oxigenación de los océanos y, en particular, las condiciones de muy bajo oxígeno que siguieron a la anomalía isotópica de Lomagundi).
- Holland, H. D., 2002. «Volcanic gases, black smokers, and the great oxidation event». Geochimica et Cosmochimica Acta 66, 3811-3826. (Dick Holland aporta aquí una posible asociación entre el GOE y la anomalía isotópica de Lomagundi, basada en la disponibilidad de fósforo). —, 2004. «The geologic history of seawater», en: Holland, H. D., K. K. Turekian (editores), Treatise on Geochemistry. Elsevier, Amsterdam, pp. 583-625. (Una revision excelente y panorámica, pero aquí en particular Holland calculó cómo la presencia de Fe2+ en las aguas superficiales a lo largo de una gran plataforma continental restringe las concentraciones atmosféricas de oxígeno).
- Planavsky, N. J., P. McGoldrick, C. T. Scott, C. Li, C. T. Reinhard, A. E. Kelly, X. Chu y col., 2011. «Widespread iron-rich conditions in the mid-Proterozoic ocean». Nature 477, 488-451. (Demostración de las generalizadas condiciones ferruginosas a lo largo del proterozoico medio).
- Poulton, S. W., D. E. Canfield, P. Fralick, 2004. «The transition to a sulfidic ocean ~1.84 billion years ago». Nature 431, 173-177. (Demostración de una transición desde formaciones de hierro bandeado hasta condiciones euxínicas tras la deposición de la formación de hierro de Gunflint). —, P. W. Fralick, D. E. Canfield, 2010. «Spatial variability in oceanic redox structure 1.8 billion years ago». Nature Geoscience 3, 486-490. (Documenta la estructura 2-D de la química oceánica durante y después de la deposición de la formación de hierro de Gunflint. Muestra que las condiciones euxínicas se extendían a unos 100 kilómetros de la costa, y desde ahí se desarrollaban las condiciones ferruginosas).
- Raiswell, R, D. E. Canfield, 2012. «The iron biogeochemical cycles past and present». Geochemical Perspectives 1, 1-20. (El reciclado bioquímico del hierro, en el pasado y ahora, siguiendo el relato de las carreras científicas de Raiswell y Canfield).
- Sarmiento, J. L., T. D. Herbert, J. R. Toggweiler, 1988. «Causes of anoxia in the world ocean». Global Biogeochemical Cycles 2, 115-128. (Modelo simple y bello de los controles de la oxigenación de las aguas oceánicas profundas).
- Shen, Y., A. H. Knoll, M. R. Walter, 2003. «Evidence for low sulphate and anoxia in a mid-Proterozoic marine basin». Nature 423, 632-635. (Mostró la presencia de prolongadas condiciones sulfurosas de las aguas profundas en una cuenca marina de aproximadamente 1500 millones de años de Northern Territory, Australia).
- Slack, J. F., T. Grenne, A. Bekker, O. J. Rouxel, P. A. Lindberg, 2007. «Suboxic deep seawater in the late Paleoproterozoic: Evidence from hematitic chert and iron formation related to seafloor-hydrothermal sulfide deposits, central Arizona, USA». Earth and Planetary Science Letters 255, 243-256. (Muestra que las aguas oceánicas profundas no eran sulfurosas en el eón paleloproterozoico tardío).
- Wilkinson, B. H., B. J. McElroy, S. E. Kesler, S. E. Peters, E. D. Rothman, 2009. «Global geologic maps are tectonic speedometers: Rates of rock cycling from area-age frequencies». Geological Society of America Bulletin 121, 760-779. (Un moderno análisis de la preservación de las rocas en función del tiempo).
CAPÍTULO 10- Billings, E., 1872. «On some fossils from the Primordial rocks of Newfoundland». The Canadian Naturalist 4. 465-479. (Primera descripción de la fauna de Ediacara).
- Bjerrum, C. J., D. E. Canfield, 2011. «Towards a quantitative understanding of the late Neoproterozoic carbon cycle». Proceedings of the National Academy of Sciences of the United States of America 108, 5542-5547. (Un modelo para explicar las grandes anomalías isotópicas negativas basándose en el funcionamiento inestable del ciclo del metano).
- Butterfield, N. J., 2011. «Animals and the invention of the Phanerozoic Earth system». Trends in Ecology and Evolution 26, 81-87. (Un enfoque de cómo los animales han dado forma al medio geoquímico).
- Canfield, D. E., S. W. Poulton, A. H. Knoll, G. M. Narbonne, G. Ross, T. Goldberg, H. Strauss, 2008. «Ferruginous conditions dominated later Neoproterozoic deep water chemistry». Science 321, 949-952. (Artículo que muestra el desarrollo generalizado de unas condiciones ferruginosas en aguas profundas durante la era neoproterozoica).
- Canfield, D. E., S. W. Poulton, G. M. Narbonne, 2007. «Late-Neoproterozoic deep-ocean oxygenation and the rise of animal life». Science 315, 92-95. (Artículo que muestra la oxigenación del océano profundo de la península de Avalon hace unos 580 millones de años).
- Dahl, T. W., E. U. Hammarlund, A, D. Anbar, D. P. G. Bond, B. C. Gill, G. W. Gordon, A. Knoll y col., 2010. «Devonian rise in atmospheric oxygen correlated to the radiations of terrestrial plants and large predatory fish». Proceedings of the National Academy of Sciences of the United States of America PNAS 107, 17911-17915. (Utiliza evidencias con isótopos del molibdeno en apoyo de un aumento neoproterozoico de las concentraciones de oxígeno atmosférico, así como de otro aumento más tardío, en el periodo devónico).
- Derry, L. A., S. B. Jacobsen, 1990. «The chemical evolution of precambrian seawater: Evidence for REEs in banded iron formation». Geochimica et Cosmochimica Acta 54, 2965-2977. (Inteligente aplicación de múltiples sistemas de isótopos para calcular la tasa de enterramiento de isótopos de carbono durante la era neoproterozoica).
- Knoll, A. H., 2011. «The multiple origins of complex multicellularity». Annual Review of Earth and Planetary Sciences 39, 217-239. (Fascinante exploración de la evolución de la multicelularidad entre diferentes grupos de organismos, con una maravillosa discusión de la evolución temprana de los animales).
- Knoll, A. H., J. M. Hayes, A. J. Kaufman, K. Swett, I. B. Lambert, 1986. «Secular variation in carbon isotope ratios from Upper Proterozoic successions of Svalbard and East Greenland». Nature 321, 832-838. (Primer registro de isótopos de carbono que indica unas altas tasas de enterramiento de carbono durante la era neoproterozoica).
- Narbonne, G. M., 2005. «The Ediacara biota: Neoproterozoic origin of animals and their ecosystems». Annual Review of Earth and Planetary Science 33, 421-442. (Excelente revision de la fauna de Ediacara).
- Nursall, J. R., 1959. «Oxygen as a prerequisite to the origin of the metazoa». Nature 183, 1170-1172. (Discusión pionera de la relación entre la historia de las concentraciones de oxígeno atmosférico y la evolución animal).
- Rothman, D. H., J. M. Hayes, R. E. Summons, 2003. «Dynamics of the Neoproterozoic carbon cycle». Proceedings of the National Academy of Sciences of the United States of America 100, 8124-8129. (Sagaz análisis de las anomalías isotópicas del carbono de la era neoproterozoica, con la insinuación de que se originaron por la oxidación periódica de un gran reservorio de carbono orgánico disuelto en el mar).
- Runnegar, B., 1982. «Oxygen requirements, biology and phylogenetic significance of the late Precambrian worm Dickinsonia, and the evolution of the burrowing habit». Alcheringa 6, 223-239. (Un intento de poner límites a los niveles de oxígeno atmosférico durante la emergencia de la vida animal, basado en cálculos sobre los requisitos fisiológicos del fósil de Ediacara Dickinsonia).
- Seilacher, A., 1992. «Vendobionta and Psammocorallia: Lost constructions of Precambrian evolution». Journal of the Geological Society of London 149, 607-613. (Propuesta de Seilacher para la afinidad biológica de la fauna de Ediacara).
- Shen, Y., T. Zhang, P. F. Hoffman, 2008. «On the coevolution of Ediacaran oceans and animals». Proceedings of the National Academy of Sciences of the United States of America 105, 7376-7381. (Muestra el desarrollo de la oxigenación de las aguas profundas hace unos 580 millones de años).
- Sahoo, S. K., N. J. Planavsky, B. Kendall, X. Wang, X. Shi, C. Scott, A. D. Anbar y col., 2012. «Ocean oxygenation in the wake of the Marinoan glaciation». Nature 489, 546-549. (Aporta evidencias de la oxigenación del océano justo después de la glaciación Marinoan, hace 630 millones de años).
- Sprigg, R. C., 1947. «Early Cambrian (?) jellyfishes from the Flinders ranges, South Australia». Transactions of the Royal Society of South Australia 71, 212-224. (El artículo que puso en el mapa a los fósiles de Ediacara).
CAPÍTULO 11- Bergman, N. M., T. M. Lenton, A. J. Watson, 2004. «COPSE: A new model of biogeochemical cycling over Phanerozoic time». American Journal of Science 304, 397-437. (Modelo de evolución del oxígeno independiente de isótopos a lo largo del fanerozoico, basado en un número limitado de «conductores» del input).
- Berner, R. A., 1987. «Models for carbon and sulfur cycles and atmospheric oxygen: Application to peleozoic geologic history». American Journal of Science 287, 177-196. (Primer intento de Bob Berner de construir un modelo de regulación del oxígeno. Se introduce la idea de reciclado rápido). —, 2006. «GEOCARBSULF: A combined model for Phanerozoic atmospheric O2 and CO2». Geochimica et Cosmochimica Acta 70, 5653-5664. (Un modelo totalmente acoplado del oxígeno, el carbono y el azufre, con numerosos outputs como el CO2 y el O2 atmosféricos y diversos componentes químicos de los océanos. La plataforma de modelos más avanzada de Bob Berner). —, D. E. Canfield, 1989. «A model for atmospheric oxygen over Phanerozoic time». American Journal of Science 289, 333-361. (Niveles de oxígeno atmosférico a partir de los datos de abundancia de rocas). —, A. C. Lasaga, R. M. Garrels, 1983. «The carbonate-silicate geochemical cycle and its effect on atmospheric carbon dioxide over the past 100 million years». American Journal of Science 283, 641-683. (Artículo clásico sobre el modelado de la historia del CO2 atmosférico).
- Butterfield, N. J., 2011. «Animals and the invention of the Phanerozoic Earth system». Trends in Ecology and Evolution 26, 81-87. (El punto de vista de Nick Butterfield sobre el papel de los animales en la formación de su entorno químico).
- Dahl, T. W., E. U. Hammarlund, 2011. «Do large predatory fish track ocean oxygenation?». Communicative & Integrative Biology 4, 1-3. (Un buen estudio de modelado que muestra cómo la tasa metabólica de los peces y su tamaño están acoplados a los niveles de oxígeno disponible). —, E. U. Hammarlund, A. D. Anbar, D. P. G. Bond, B. C. Gill, G. W. Gordon, A. H. Knoll y col., 2010. «Devonian rise in atmospheric oxygen correlated to the radiations of terrestrial plants and large predatory fish». Proceedings of the National Academy of Sciences of the United States of America 107, 17911-17915. (Presenta evidencias con isótopos del molibdeno de un aumento del oxígeno atmosférico durante el periodo devónico).
- Garrels, R. M., A. Lerman, 1981. «Phanerozoic cycles of sedimentary carbon and sulfur». Proceedings of the National Academy of Sciences of the United States of America 78, 4652-4656. (El artículo con el que comenzó todo. Un modelo revelador de la evolución de los ciclos del carbono y el azufre a lo largo del fanerozoico). —, F. T. Mackenzie, 1971. Evolution of Sedimentary Rocks. W.W. Norton, Nueva York. (Un verdadero clásico. Envuelve el recién hallado reconocimiento de la tectónica de placas con percepciones fundamentales de la composición y el reciclado geológico de las rocas sedimentarias).
- Harlé, É., A. Harlé, 1911. «Le vol de grands reptiles et insectes disparus semble indiquer une pression atmosphérique élevée». Bulletin de la Société Géologique de France 11, 118-121. (Una de las primeras discusiones, tal vez la primera, de los niveles de oxígeno atmosférico y el gigantismo de los insectos).
- Harrison, J. F., A. Kaiser, J. M. VandenBrooks, 2010. «Atmospheric oxygen level and the evolution of insect body size». Proceedings of the Royal Society of London B 277, 1937-1946. (Buena revisión del modo en que el tamaño de los insectos se ve influido por los niveles de oxígeno en sistemas experimentales).
- Kump, L. R., Garrels, R. M., 1986. «Modeling atmospheric O2 in the global sedimentary redox cycle». American Journal of Science 286, 337-360. (El primer modelo completo de la evolución del oxígeno atmosférico a lo largo del tiempo geológico. Basado en la filosofía de modelado de Garrels y Lerman).
- Lamsdell, J. C., S. J. Braddy, 2010. «Cope’s Rule and Romer’s theory: Patterns of diversity and gigantism in eurypterids and Palaeozoic vertebrates». Biology Letters 6, 265-269. (Buena exploración de la historia de la evolución del tamaño entre los euriptéridos, que se asocia a la evolución de los niveles de oxígeno atmosférico).
- Ward, P. D., 2006. Out of Thin Air. Joseph Henry Press, Washington. (Una exploración del modo en que los cambios del oxígeno atmosférico han influido en la evolución animal durante el eón fanerozoico. Basado con fuerza en el modelo Geocarbsulf).
Epílogo - Kump, L. R., 2008. «The rise of atmospheric oxygen». Nature 451, 277-278. (Breve y excelente revisión de la historia del oxígeno atmosférico).
Alfombra de bacterias oxidantes del sulfuro en sedimentos hidrotermales de la cuenca de Guaymas, golfo de México. Foto tomada mientras el autor estaba a bordo del Alvin.

Poblaciones microbianas fotótrofas estratificadas en arena de playa de la isla de Bornholm, Dinamarca. Foto del autor.

Una formación de hierro bandeado (BIF) de 2500 millones de años en la garganta de Dales, Australia Occidental. Foto del autor.
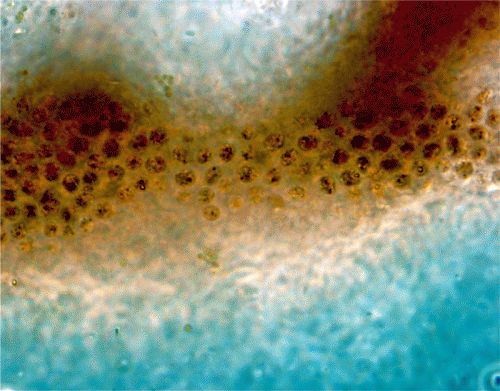
Población natural de bacterias púrpuras del azufre. Estos organismos concretos están implicados probablemente en la oxidación del sulfuro, pero tienen una relación estrecha con las bacterias púrpuras fotótrofas oxidantes del hierro. Foto cortesía de Bo Thamdrup y Jakob Zopfi.
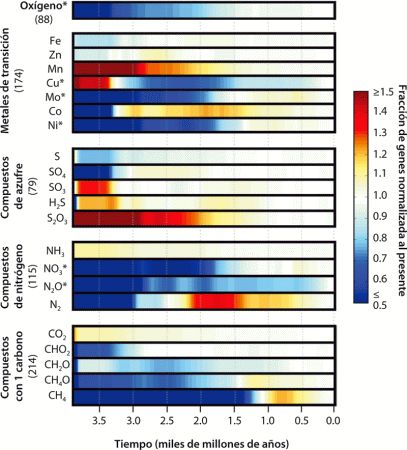
Evolución del uso de compuestos sensibles a redox (reacciones de reducción/oxidación) en el metabolismo microbiano. Los colores indican la abundancia de enzimas que se unen a cada compuesto con relación al presente. Siguiendo este esquema, la utilización de oxígeno, y de diversos compuestos del nitrógeno y compuestos C1 (compuestos con un solo átomo de carbono, incluido el metano CH4), evolucionó bastante tarde, mientras que la utilización de compuestos de manganeso y azufre evolucionó pronto. Ligeramente modificado a partir de David y Alm (2011), con permiso.
Comparación de las estructuras de las proteínas del centro de reacción fotosintético. Versión en color de la figura 3.4.

Alfombra de cianobacterias estratificada de Guerrero Negro, península de Baja, México. Foto del autor.

Estromatolito moderno de la bahía de Shark, Australia. Foto del autor.

Peñasco rojo brillante de la formación Gaskier, comparada con la coloración gris apagada del peñasco adyacente de la superpuesta formación Drook. Foto del autor.
Notas: [1] La navaja de Ockham se atribuye al fraile inglés del siglo XIV Guillermo de Ockham. En su forma original, traducida del latín, dice: «No se deben multiplicar las entidades innecesariamente». En términos más ordinarios, la navaja de Ockham enuncia que la mejor explicación de algo suele ser la más simple.
[2] Las reacciones redox deben ser favorables termodinámicamente y, para el aficionado, los organismos realmente requieren que las reacciones sean más que meramente favorables termodinámicamente. De hecho, la reacción debe ser favorable por unos 15 o 20 kilojulios por mol de carbono orgánico oxidado durante el metabolismo heterótrofo. Esto se debe a que, al nivel celular, la función biológica más básica es la producción de ATP. Esto requiere la traslocación de 3 a 4 protones (H
+) a través del un complejo enzimático conocido como ATPasa (pronunciado
atepeasa), y la energía mínima que necesita un organismo, por lo tanto, es la energía requerida para traslocar un protón, que se estima en 15 o 20 kilojulios por mol de carbono orgánico oxidado.
[3] Cuando las condiciones se ponen difíciles, y/o el agua se vuelve escasa, muchos organismos pueden formar esporas o quistes y permanecer en una especie de animación suspendida casi indefinidamente, hasta que las condiciones mejoran y pueden reiniciar el metabolismo.
[4] El nombre
zona Goldilocks remite al cuento tradicional escocés
Goldilocks y los tres osos, o
Ricitos de Oro y los tres osos, en el que la niña Goldilocks se topa con la casa de los tres osos. La casa está vacía y, al entrar, Goldilocks ve que la crema de avena del osito pequeño, su silla y su cama son «justo las adecuadas» para ella, y luego se duerme en esa camita. Cuando los tres osos regresan, el osito despierta a Goldilocks y ella salta deprisa por la ventana para escapar de la casa.
[5] El albedo define la reflectividad de un objeto a la luz visible. Un objeto con un albedo cero es perfectamente negro y absorbe toda la luz que le llega, mientras que un cuerpo perfectamente reflectivo es blanco, con albedo 1. Así, un objeto de bajo albedo absorberá más energía lumínica y se calentará más que un objeto de alto albedo. El albedo de la Tierra se estima en 0,3, incluyendo el efecto de las nubes.
[6] La atmósfera actual de Venus contiene muy poca agua. La mayor parte del agua que tenía originalmente se perdió por fotolisis, un proceso estimulado por la luz que produce oxígeno gaseoso e hidrógeno gaseoso. En Venus, el hidrógeno debió de escapar al espacio, mientras que el oxígeno habría reaccionado con los minerales de la superficie del planeta, o bien con gases procedentes del interior. De hecho, la atmósfera actual de Venus es rica en CO
2, ya que el CO
2 se ha acumulado durante eones a medida que salía del interior del planeta.
[7] El Sol brilla más cada vez conforme la proporción H
2/He en su núcleo decrece por las reacciones de fusión nuclear. Según el
modelo solar estándar, esto conduce a una contracción gravitatoria, la liberación de calor y un incremento de temperatura, que se expresa como un incremento de luminosidad. Se estima que, cuando el Sol se acababa de formar, hace unos 4500 millones de años, sólo tenía el 70% de la luminosidad actual.
[8] Soy del medio oeste de Estados Unidos, donde las lápidas se suelen tallar en caliza. Las lápidas con más de 150 años suelen ser ilegibles, o casi. La razón es el proceso de erosión. El CO
2 de la atmósfera se disuelve en la lluvia y reacciona con la caliza, corroyéndola lentamente a medida que la lluvia corre por su superficie.
[9] Estas evidencias vienen de la distribución de minerales en los suelos antiguos (de 2700 a 2200 millones de años atrás), conocidos como paleosuelos. Estos paleosuelos carecen de siderita, lo que, según los cálculos de Rob Rye, Phillip Kuo y Dick Holland, significa que los niveles atmosféricos de CO
2 eran mucho menores que los esperados (Rye y colaboradores, 1995. Las concentraciones atmosféricas de dióxido de carbono antes de 2200 millones de años atrás.
Nature 378, 603-605). Este tópico se discute en detalle en el capítulo 7.
[10] Unos altos niveles de metano pueden encajar también con nuestra comprensión de la manera en que las concentraciones de oxígeno han cambiado con el tiempo, como se revelará en capítulos posteriores; el oxígeno y el metano son termodinámicamente inestables si están juntos, y reaccionan en la atmósfera formando CO
2.
[11] Minik Rosing y sus colegas arguyeron que el albedo de la Tierra joven era menor de lo que se asume normalmente, porque el área continental era menor que en el presente, y porque se habrían generado menos nubes debido a un menor número de núcleos de condensación inducidos biológicamente. Con un menor albedo, no se requeriría tanta concentración de gases de efecto invernadero en la atmósfera para templar la Tierra, incluso con un Sol joven menos brillante. Estos investigadores también presentaron evidencias adicionales en apoyo de una atmósfera baja en CO
2, argumentando que la abundancia de magnetita en las formaciones de hierro bandeado que datan del eón arcaico es incoherente con unos altos niveles de CO
2 atmosférico en ese periodo. Si el CO
2 hubiera sido mucho más alto que ahora, la siderita sería la fase dominante de Fe (hierro).
[12] En la atmósfera actual hay 3,16×10
15 Kg de CO
2 (a 400 ppm, o partes por millón), lo que equivale a 72 ×10
15 moles. Las tasas totales de fotosíntesis en la Tierra ascienden a unos 8 ×10
15 moles/año, así que el tiempo de residencia del CO
2 en la atmósfera con respecto a su retirada por fotosíntesis es de 72 ×10
15 moles/(8 ×10
15 moles/año), o unos 9 años.
[13] Hay en la actualidad 3,3 ×10
15 moles de fosfato en los océanos. Las tasas marinas de fotosíntesis ascienden a unos 4 ×10
15 moles/año, y la fracción media de C/P (carbono a fósforo) del fitoplancton es 106:1, de modo que el tiempo de residencia del P con respecto a su captación por el plancton es [3,4 ×10
15 moles/(4 ×10
15 moles/año)] ×106, o unos 86 años.
[14] Mis colegas geoquímicos me pondrán objeciones aquí, porque, como se menciona en el texto, el carbono también es retirado permanentemente en los sedimentos como minerales de carbonato inorgánico. Además, el bicarbonato es la principal forma de carbono inorgánico en los océanos y su reservorio supone unas 50 veces más que el CO
2 en la atmósfera. El resultado final de todo esto sería alargar el tiempo de residencia calculado en un factor de 10, más o menos. Tener en cuenta estas consideraciones, sin embargo, no cambiará el argumento que se hace en el texto.
[15] De niño tenía una pesadilla recurrente en la que gateaba por una cueva hasta llegar a una angostura en que me quedaba atascado. Espeluznante.
[16] El agua del río Colorado se utiliza hasta la extenuación para regar, y es una fuente importante de agua potable para el sur de California. Como resultado, al golfo de California no llaga más que un hilillo de agua.
[17] Esto se suele llamar la «zona fótica».
[18] Son miembros del filo
Annelida, y por tanto primos lejanos de la lombriz de tierra y la sanguijuela.
[19] Los organismos autótrofos, incluidos los autótrofos metanógenos, obtienen su material celular del CO
2.
[20] La reacción es 4H
2 + CO
2 CH
4 + 2H
2O.
[21] Los heterótrofos en general obtienen su material celular de la materia orgánica. Los metanógenos heterótrofos, en particular, están especializados en romper el acetato en metano y CO
2 (la reacción es H
+ + CH
2COO
-à CH
4 + CO
2). El acetato es un producto de fermentación común. Algunos metanógenos pueden también descomponer compuestos de metilamina, liberando metano, y también pueden producir metano a partir de alcoholes simples como el metanol.
[22] Las reacciones son:
SO42—+ 2CH2O → H2S + 2HCO3—
SO42—+ 4H2 + 2H+ → H2S + 4H2O
[23] El H
2 es un compuesto químico altamente reducido, y por tanto muy adecuado para reducir el CO
2 a materia orgánica, y el sulfato a sulfuro, y también puede reducir muchas otras especies químicas oxidadas. Recordemos del capítulo anterior que los organismos consiguen energía y crecen controlando estas reacciones de oxidación-reducción termodinámicamente favorables.
[24] Esto significa, literalmente, organismos fotosintéticos no productores de oxígeno.
[25] La reacción es: 2H
2 + CO
2 → H
2O + CH
2O.
[26] En referencia a los organismos procariotas, el famoso geobiólogo Laurens Baas Backing enunció en su libro de 1934
Geobiología: «Todo está en todas partes, pero el entorno selecciona». Pienso que Fritz Widdel es un firme creyente en ese principio. Nunca parece viajar muy lejos del laboratorio para recolectar algunas de las especies microbianas más interesantes.
[27] Para compartir algunas, al hacer nuestros cálculos asumimos que la disponibilidad de nutrientes y la velocidad de circulación de los océanos eran las mismas que hoy.
[28] El nombre es una broma, porque este es un lugar increíblemente caliente.
[29] Esta cita y las siguientes de Scheele están tomadas como extractos traducidos del volumen original de Scheele. Los extractos son de
El descubrimiento del oxígeno, parte 2, Experimentos de Carl Wilhelm Scheele (1777), Alembic Club Reprints, no. 8, Edimburgo (1901), traducción de Leonard Dobbin.
[30] El alquimista e inventor Comelis Drebbel (1572-1633) descubrió, aparentemente, que el «salpetre aéreo» (oxígeno) respirable podía producirse calentando nitrato potásico, y supuestamente usó esta técnica en 1621 para sustentar la regata de 12 remeros durante 10 millas bajo el agua, en lo que constituyó probablemente el primer viaje submarino sostenido.
[31] Realmente el 21% de nuestra atmósfera.
[32] Priestley se refirió al oxígeno como
aire deflogistado, y lo siguió haciendo hasta su muerte en 1804. La lógica que subyacía a este nombre era que, si la acumulación de flogisto procedente de sustancias en combustión causaba que el aire perdiera su capacidad de sustentar la llama, el aire deflogistado tendría la propiedad opuesta y sustentaría la llama.
[33] Esta posible cadena de sucesos se explora en la obra de teatro
Oxígeno de Carl Djerassi, de la Universidad de Stanford, y Roald Hoffmann, de la Universidad de Cornell.
[34] Esta cita es de Jan Ingenuos, 1779,
Experimentos con vegetales, descubriendo su gran poder de purificar el aire común a la luz del sol, y de dañarlo a la sombra y por la noche. A lo que se suma un nuevo método de determinar el grado exacto de salubridad de la atmósfera. Impreso por P. Elmsly en el Strand y H. Payne en Pall Mall, Londres.
[35] El transportador de electrones soluble NADP
+ es un compuesto oxidado que acepta electrones para formar NADPH, su forma reducida complementaria. El par redox NADP
+/NADPH, y el par relacionado NAD
+/NADH, se usan exhaustivamente por las células para conducir reacciones de oxidación-reducción durante el metabolismo y la biosíntesis.
[36] ATP significa adenosín trifosfato (o trifosfato de adenosina). Es un compuesto de alta energía usado por las células para conducir reacciones químicas que de otro modo serían termodinámicamente imposibles. Una forma importante de producción de ATP es mediante la llamada cadena de transporte de electrones, en la que la transferencia de electrones a través de una serie de enzimas transportadoras se acopla al transporte de protones a través de una membrana. Los protones y los electrones fluyen de vuelta a través de una enzima conocida como ATP sintasa, formando ATP en el proceso.
[37] Rubisco es una abreviatura de ribulosa 1,5-bifosfato carboxilasa/oxigenasa.
[38] Una pequeña digresión sobre las GSB (bacterias verdes del azufre). Como norma, las GSB huyen del oxígeno, y la mayoría son expertas en oxidar el sulfuro, aunque algunas pueden también oxidar el Fe2
+ (se encuentran en el lago Matano, rico en Fe2
+, que vimos en el capítulo 2). Algunas GSB han desarrollado también enormes complejos antena, y viven en unas condiciones de luz absurdamente baja. Por ejemplo, en el mar Negro, las GSB oxidan el sulfuro a unas profundidades de 100 metros. Aquí, la luz es equivalente a la que tendríamos en una noche sin luna ni nubes en el desierto del Mohave ¡llevando dos pares de gafas de sol razonablemente gruesas! Inténtalo alguna vez.
[39] Los fotótrofos anoxigénicos miembros de este grupo son muy diversos en estilo de vida, y viven en entornos que abarcan desde plantas de tratamiento de aguas residuales hasta lagos sulfurosos. Muchos de estos organismos también pueden oxidar el sulfuro, y algunos pueden oxidar el Fe2
+ también. Como grupo, sin embargo, son generalistas, y algunos miembros pueden encontrarse también en entornos bien oxigenados, incluidas las capas altas e iluminadas por el sol del océano.
[40] Las duplicaciones de genes no son infrecuentes en la historia de la vida. Puede ocurrir toda una gama de duplicaciones, desde genes individuales hasta genomas enteros. Un gen duplicado, sin embargo, puede evolucionar mediante mutaciones de forma independiente al gen original del que se duplicó. En algunos casos (la mayoría, probablemente), los genes duplicados se vuelven inservibles y simplemente desaparecen del genoma. En otros casos, el gen duplicado puede compartir la función del gen original, y puede incluso acabar funcionando mejor que la versión original. Todavía en otros casos, un gen duplicado puede evolucionar para llevar a cabo una función enteramente nueva. En general, si el gen duplicado sigue siendo beneficioso para la bacteria, se retendrá en el genoma, y si no lo es, se perderá.
[41] De hecho, todas las Rubisco tienen una actividad oxigenasa, incluidas las llamadas proteínas tipo Rubisco (RLP), asociadas con algunas arqueas anaeróbicas. Estas RLP no se usan para fijar carbono, pero se cree que son las precursoras de las verdaderas Rubisco fijadoras de carbono.
[42] Una posible ventaja de Rubisco es que, aunque tiene actividad oxigenasa, aún puede funcionar con el ciclo de Calvin en presencia de oxígeno, lo que no es el caso de otras rutas de fijación del carbono.
[43] Cuando las cianobacterias forman alfombras microbianas, las concentraciones de oxígeno dentro de las alfombras pueden alcanzar niveles de 1 bario o más altas, como veremos en el capítulo 4. Esto debió de ser cierto también hace mucho tiempo, cuando los niveles de oxígeno atmosférico eran mucho más bajos que hoy.
[44] La producción primaria se refiere a la tasa de formación de materia orgánica a partir del CO
2 mediante procesos autótrofos. Es habitual referirse ya sea a la producción primaria bruta (GPP,
gross primary production) o a la producción primaria neta (NPP,
net primary production). La producción primaria bruta se refiere a la velocidad instantánea de fijación de CO
2 en compuestos orgánicos, mientras que la neta se refiere a la velocidad de producción de materia orgánica una vez que se sustrae la respiración celular: NPP = GPP — respiración.
[45] Por supuesto, la Tierra se volvió verde realmente con la muy, muy posterior evolución de las plantas terrestres.
[46] Al menos, se habría requerido un microscopio en cualquier zona menos en las fuentes hidrotermales terrestres, donde las comunidades fotosintéticas anoxigénicas pueden haber sido bien visibles, como vimos en el capítulo 2.
[47] En este caso, los depósitos consistían en poco más que una casucha de madera donde se podía comprar cerveza, soda y (a veces) hielo. La cerveza era barata, el servicio amigable, y siempre había dos o tres perros ganduleando por la basura, situada justo enfrente de la puerta.
[48] Los estromatolitos marinos modernos que quizá sean más famosos se encuentran es Shark Bay, en Hamlin Pool, Australia, donde la salinidad alcanza más o menos el doble de la del agua marina normal. Esta agua salada supuestamente disuade a los animales de pasto, que de otra manera se comerían a las florecientes cianobacterias. En general, los estromatolitos se forman a medida que las sustancias pegajosas producidas por las cianobacterias atrapan y adhieren partículas sólidas, o precipitan carbonato cálcico del agua del mar, formando estructuras sólidas parecidas a montículos.
[49] Esto ha sido medido realmente por Bo Barrer Jorgensen, de la Universidad de Aarhus en Dinamarca. Como un maestro de la investigación sobre las alfombras microbianas, Bo desarrolló unas sondas que le permitieron explorar la intensidad y la distribución espectral de la luz en las alfombras microbianas con una resolución de 0,1 mm de profundidad.
[50] Niels Peter Revsbech desarrolló un método llamado cambio de luz-oscuridad para determinar las tasas de producción de oxígeno. La premisa es muy hábil, y en realidad muy simple. Cuando el perfil de oxígeno se encuentra en estado estacionario, o que no cambia con el tiempo, en cualquier punto del perfil la tasa de producción de oxígeno está equilibrada por la tasa de pérdida de oxígeno por respiración y difusión. Niels Peter razonó que, si apagaba las luces, eliminaría la producción de oxígeno, pero no los procesos responsables de su retirada. Por tanto, justo después de apagar la luz, la velocidad inmediata de descenso del oxígeno medida con el microelectrodo es exactamente la misma que la velocidad de producción de oxígeno justo antes de que se apagara la luz. En la práctica, para medir la tasa de producción de oxígeno (equivalente a la tasa de producción primaria, véase nota 8 más abajo), una alfombra microbiana se pone a oscuras y la velocidad de descenso del oxígeno se mide durante un segundo o dos. Después de esto, el perfil de oxígeno empieza a cambiar de forma, y la velocidad de descenso del oxígeno ya no equivale a la velocidad de producción de oxígeno antes de quedarse a oscuras.
[51] Las tasas de producción de materia orgánica y de producción de oxígeno por fotosíntesis oxigénica son aproximadamente equivalentes. Esto se puede ver en la ecuación de la fotosíntesis: CO
2 + H
2O → CH
2O + O
2, donde CH
2O representa materia orgánica.
[52] Los procariotas son lo que normalmente llamamos bacterias. Son organismos unicelulares que carecen, salvo en raros casos, de orgánulos y un núcleo envuelto en membranas. Formalmente se dividen en bacterias y arqueas, dos dominios del árbol de la vida.
[53] Este uso de la energía lumínica para utilizar oxígeno se llama «reacción de Mehler».
[54] El oxígeno también supone el 21% de los gases atmosféricos totales en la cima del monte Everest, donde la baja concentración de oxígeno (por volumen de gas) se debe a la baja presión atmosférica.
[55] Tal vez habría que añadir un poco de CO
2 al recipiente, de manera que no empiece a limitar la producción de oxígeno.
[56] Las plantas respiran, al igual que nosotros, para obtener energía que alimente sus funciones metabólicas. Hacen esto noche y día, pero de noche no hay producción fotosintética de carbono, y por tanto hay una oxidación de carbono neta. El grado hasta el que el oxígeno se consume por la noche variará mucho de planta a planta, dependiendo sobre todo de la velocidad de crecimiento de la planta. Las plantas de crecimiento rápido permitirán más acumulación de oxígeno que las plantas de crecimiento lento.
[57] La materia orgánica podría incluir restos de la materia orgánica original producida fotosintéticamente, o cualquiera de los organismos que se han alimentado de ella cadena trófica arriba. Esto incluiría casi cualquier organismo no fotosintético desde las bacterias heterótrofas hasta los hongos, los peces y los caballos.
[58] Una pizarra es una roca sedimentaria de grano fino, pobre en carbonato y compuesta predominantemente de arcilla y partículas con la textura del limo.
[59] Estas son las ecuaciones relevantes que muestran cómo el enterramiento de la pirita representa una fuente de oxígeno para la atmósfera. La última ecuación es la suma de las otras tres.
16H+ + 16HCO3- → 16CH2O + 16O2 (fotosíntesis oxigénica)
8SO42— + 16CH2O → 16HCO3— + 8H2S (reducción del sulfato)
8H2S + 2Fe2O3 + O2 → 8H2O + 4FeS2 (formación de pirita)
16H+ + 8SO42— + 2Fe2O3 → 8H2O + 4FeS2 + 15O2 (suma)
[60] Tristemente, hoy ambos están muertos. Bob Garrels falleció en 1988, mientras que Karl Turekian murió justo antes de que yo recibiera las galeradas de este libro para su corrección. Los dos fueron gigantes de la geoquímica e importantes mentores para mí en las fases iniciales de mi desarrollo como científico.
[61] Por supuesto, no todos los sedimentos serían propensos a este tipo de erosión. Se supone que una fracción del sedimento recién depositado se hace disponible para la erosión en una escala de tiempo corta (algo así como 10 o 20 millones de años), mientras que el resto se hace parte del
viejo registro de rocas de reciclado más lento.
[62] La subducción es una consecuencia natural del proceso de tectónica de placas por el que el suelo marino creado originalmente en las dorsales centro-oceánicas es subducido de vuelta al interior del manto. Las áreas de subducción son zonas de construcción de montañas y gran actividad volcánica. La costa occidental de Suramérica es un buen ejemplo de zona de subducción.
[63] Anóxico significa sin oxígeno, y óxico, con oxígeno. La anoxia oceánica se refiere a una condición en que partes de la columna de agua del océano no contienen oxígeno. El grado de anoxia oceánica se ha expandido y contraído a lo largo de la historia de la Tierra en formas que se discuten por todo el texto.
[64] En los inicios de mi carrera, me impliqué bastante en estudiar los procesos que controlan la preservación del carbono orgánico en los sedimentos, y si la preservación se reforzaba durante la descomposición anóxica. Como suele ocurrir, esa investigación fue un poco complicada, pero las evidencias empíricas aportadas por mí entre otros, y los estudios experimentales posteriores, en particular de mi colega Eric Kristensen, de la Universidad del Sur de Dinamarca, muestran que la materia orgánica se descompone más a fondo en presencia de oxígeno que por los procesos anaeróbicos que operan en su ausencia.
[65] Hay en verdad mucho debate sobre la importancia relativa del nitrógeno y el fósforo en el control de las tasas de producción primaria en los océanos. Si uno mira a las tendencias de N y de P en el océano global, en la mayoría de los sitios hay un ligero exceso de fósforo; esto implica que el nitrógeno es el nutriente limitante, y por tanto el que es más probable que controle las tasas de producción primaria. El ligero déficit de nitrógeno surge porque la tasa de fijación de nitrógeno no está a la altura de la tasa de desnitrificación en el océano. Este puede considerarse el punto de vista más
biológico sobre la limitación de nutrientes. Los geoquímicos tienden a argumentar que el fósforo es el nutriente limitante porque el contenido de nitrógeno de los océanos debería ajustarse al contenido de fósforo con respecto a las necesidades del fitoplancton que usa los nutrientes. Por ejemplo, si las existencias de fosfato de los océanos se duplicaran, uno podría predecir que la fijación de nitrógeno incrementaría las existencias de nitrógeno hasta compensar (o casi) las necesidades de los productores primarios respecto al tamaño de las nuevas y mayores existencias de fósforo. Es un punto de vista digno, y por tanto hay margen para una discusión científica saludable.
[66] Las razones de las diferencias en la concentración de fósforo entre los sedimentos depositados en aguas anóxicas y oxigenadas no están completamente claras, pero se han avanzado algunas ideas. Cuando la columna de agua está oxigenada, se forman óxidos de hierro en la superficie del sedimento, y estos se adhieren con fuerza al fosfato, creando una trampa de fósforo dentro de los sedimentos. Esto podría ser parte de la razón de que los sedimentos bajo una columna oxigenada de agua contengan más fósforo. Pero no es la única razón, porque también hay más fósforo orgánico para una cantidad dada de carbono orgánico en los sedimentos depositados bajo columnas oxigenadas, en comparación con los depositados en condiciones euxínicas. Ellery Ingall ha insinuado que esto surge porque los microbios aeróbicos pueden concentrar fósforo preferentemente en su biomasa, y en fases orgánicas de fósforo que son difíciles de descomponer, en comparación con el fósforo contenido en los microbios anaeróbicos.
[67] Aunque la retroalimentación por incendios tiene sus raíces en el trabajo de Watson, a quien se suele acreditar por ello, él enunció en un comentario sobre una réplica a su estudio: «No estamos insinuando, desde luego, que el consumo de oxígeno que ocurre durante la combustión tenga relevancia alguna para el problema de la regulación del O
2. Lo que señalamos es, simplemente, que una concentración de oxígeno mucho más alta que el 21% actual sería incompatible con la existencia de una gran biomasa en tierra firme, debido a la alta probabilidad de incendios» (Watson, Lovelock, Margulis, 1980. «Discusión, lo que controla el oxígeno atmosférico».
Biosystems 12, 124-125). La posible regulación del oxígeno por incendios forestales fue expresada formalmente por primera vez por Lee Kump (Kump, 1988. «Retroalimentación terrestre en la regulación del oxígeno atmosférico por el fuego y el fósforo».
Nature 335, 152-154). Nick Lane, en su maravilloso libro (
Oxígeno: la molécula que hizo el mundo, Oxford University Press, Oxford, 2002), plantea una objeción interesante. Argumenta que el carbón producido por los incendios sería muy difícil de descomponer, y realmente podría incrementar el enterramiento de carbono orgánico, produciendo más oxígeno.
[68] Esta atribución se encuentra en el
Metalogicon de Juan de Salisbury, escrito alrededor de 1159.
[69] El Instituto Vernadsky de Geoquímica y Química Analítica de la Academia Rusa de las Ciencias está en Moscú, como también el Museo Geológico Vernadsky; en Ucrania y Rusia se pueden ver varios bustos y estatuas con su figura.
[70] Algunas ideas, como la de Ebelman, simplemente se adelantaron demasiado a su tiempo. En este caso, la idea era buena, tenía sentido, pero el campo no estaba lo bastante avanzado para hacer algo con ella. No había el suficiente contexto geológico en el que situarla, y por tanto sus implicaciones no estaban claras. Por tanto, se perdió de la conciencia científica colectiva. Fue sólo mucho después, cuando la historia geológica de la Tierra se comprendió mejor, que la idea del control del oxígeno adquirió un amplio interés entre la comunidad científica. Esta vez sí prendió al ser introducida por Garrels, Perry y Holland.
[71] Uno es el volumen 19 de la serie Scope, 1983,
El ciclo del azufre biogeoquímico global, editado por Ivanov y Freney.
[72] Aunque las rocas sedimentarias de este periodo se han perdido, se pueden encontrar algunos minerales muy primitivos. Por ejemplo, un tipo concreto de mineral conocido como circón o zircón (ZrSiO
4) se ha encontrado en algunas de las más antiguas rocas sedimentarias conocidas, y algunos de estos circones se han datado de 4300 a 4400 millones de años atrás. Las rocas que albergaban estos circones originalmente se han perdido por erosión, pero los circones en sí mismos son muy resistentes a la erosión. De este modo, se liberaron de sus rocas parentales mediante la erosión, fueron transportados por los ríos de la antigüedad y se depositaron en sedimentos más jóvenes. Estos circones contienen algunas pistas sobre la presencia o ausencia de agua en aquel planeta muy joven, y tal vez otras pistas que aún tenemos que aprender a leer.
[73] Esta redistribución es un asunto del que debemos ser conscientes en todas las rocas a las que miramos.
[74] Las turbiedades son un tipo común de sedimento, formado en general en aguas profundas. Se forman a partir de lo que se podría ver como una avalancha submarina cuando los sedimentos de profundidades más someras se removilizan y fluyen pendiente abajo hasta aguas profundas. La removilización puede ocurrir a causa de terremotos, por ejemplo, o de la inestabilidad inherente de los sedimentos que se depositan en una pendiente acusada.
[75] Hace muchos años, antes de que Minik estudiara sus rocas, Manfred Schidlowski, del Instituto Max Planck de Química en Mainz, Alemania, examinó y midió la composición isotópica del grafito en las rocas de Isua. Estas, sin embargo, no eran las mismas rocas analizadas por Minik, y no tenían el mismo contexto geológico. Sus valores estaban en general menos reducidos en
13C que los determinados por Minik (véase el texto principal para una explicación de estos isótopos del carbono).
[76] δ
13C = 1.000(R13/12
muestra - R13/12
estándar)/( R13/12
estándar), donde R13/12 es la razón de carbono-13 a carbono-12 ya sea en nuestra muestra o en nuestro control estándar.
[77] «Por mil» es lo mismo que partes por mil. Por comparación, «por ciento» (%) es lo mismo que partes por centenar. Por tanto, el valor por mil es 10 veces mayor que el valor por ciento.
[78] Hay formas de producir materia orgánica de formas no biológicas, y generan unas señales isotópicas similares a las halladas en Isua. Sin embargo, estas reacciones requieren catalizadores que no son aparentes en las rocas de Isua. Más aún, estas rutas inorgánicas, que se desconocen en la columna de agua, no pueden dar cuenta fácilmente del entreverado de estratos orgánicos con turbiedades de las rocas de Isua.
[79] Algunas cianobacterias fabrican unas células llamadas heterocistos, en las que ocurre la fijación de nitrógeno que vimos en el capítulo 4; otras cianobacterias producen células quiescentes llamadas acinetos. Tanto los acinetos como los heterocistos tienen morfologías distintivas y, si se encuentran en un filamento, aportan una evidencia convincente de cianobacterias. En otros casos, la morfología de las cianobacterias antiguas y bien preservadas se asemeja tanto a la de las modernas que interpretarlas como cianobacterias es difícil de eludir.
[80] Algunos de ellos serían bacterias del azufre filamentosas de los géneros
Beggiatoa y
Thioploca, que oxidan el sulfuro con oxígeno y nitrato para sobrevivir.
[81] Muy recientemente, Martin Brasier y sus colegas han presentado algunas evidencias de bacterias fósiles en unas rocas 60 millones de años más jóvenes que el Apex Chert (Wacey, Kilbum, Saunders, Cliff y Brasier, 2011. «Microfósiles de células metabolizadoras de azufre en rocas de 3400 millones de años en Australia Occidental».
Nature Geoscience 4, 698-702). Estas formas fósiles tienen firmas de isótopos de carbono coherentes con la vida. Los fósiles en sí mismos no están tremendamente bien conservados, y probablemente serán sometidos al mismo tipo de escrutinio que los fósiles del Apex Chert descritos por Bill Schopf, pero la prueba de los isótopos del carbono parace un indicador de vida muy sólido.
[82] También encontraron una variedad de los llamados hopanoides, o moléculas similares al hopano, y en particular 2-metil hopanos. Solía pensarse que eran biomarcadores específicos de cianobacterias, pero trabajos subsiguientes han mostrado que también son producidos por procariotas no cianobacterianos (Welander, Coleman, Sessions, Summons y Newman, 2010. Identificación de una metilasa requerida para la producción de 2-metil hopanoide e implicaciones para la interpretación de los hopanos sedimentarios.
PNAS 107, 8537-8542). En consecuencia, ya no se los considera biomarcadores estrictamente cianobacterianos.
[83] Los eucariotas son organismos cuyas células contienen un núcleo envuelto en membranas, y también orgánulos como las mitocondrias, y en plantas los cloroplastos.
[84] Tristemente, Dick falleció después de que yo escribiera la primera versión de este capítulo. Sin embargo, yo le había enviado una copia en cuanto el borrador estuvo listo, y todo lo que puedo esperar es que le gustaran las primeras páginas.
[85] El paleozoico es la primera de las tres eras del eón fanerozoico, o la edad de los animales. El eón fanerozoico se extiende desde el final del eón proterozoico, hace 542 millones de años, hasta el final del periodo pérmico, hace 251 millones de años. El final del periodo pérmico viene marcado por la mayor extinción conocida de la vida animal en la historia de la Tierra. Véase el prefacio y la figura P.1 para más detalles.
[86] De hecho, gran parte del oro se redistribuyó debido a una actividad hidrotermal posterior y, aunque aún hay un debate activo, una escuela de pensamiento mantiene que el oro redistribuido se derivó de granos de oro originales de los detritos. Parece haber algunas evidencias para esto, pues algunos granos de oro residuales son aún aparentes.
[87] Podríamos hacer excursiones a varios lugares de Estados Unidos, Canadá, Sudáfrica, India, Ucrania, Brasil y Groenlandia y observar rocas similares con edades que abarcan el eón arcaico. También hay algunos ejemplos más recientes de este tipo de rocas, de unas edades concentradas alrededor de 1900 millones de años y de 600 a 700 millones de años atrás. Las discutiremos en capítulos posteriores.
[88] James Farquhar es muy modesto, y estoy seguro de que sus palabras no fueron esas.
[89] El isótopo
32S tiene 16 protones y 16 neutrones, mientras que
33S tiene el mismo número de protones y 17 neutrones. El isótopo
34S tiene 18 neutrones, y el
36S tiene 20.
[90] Debido a la baja concentración de sulfato, no se forma yeso mientras se concentra el agua marina, así que no tenemos que preocuparnos por el yeso como una fuente de sulfato fluvial en un mundo de bajo oxígeno.
[91] Hablamos aquí del estado redox del manto. Este puede evaluarse, por ejemplo, midiendo la concentración de vanadio en las rocas volcánicas. El vanadio se reparte en una fusión parcial de rocas del manto según el estado redox del manto donde la roca fundida se formó. Por tanto, cuando estas fusiones emergen en la superficie de la Tierra, los contenidos de vanadio aportan una medida del estado redox del manto. Si examinamos esas rocas volcánicas a lo largo del tiempo, podemos evaluar la evolución del estado redox del manto, y la evidencia disponible indica que no ha variado mucho (véase por ejemplo Canil, 2002. «Vanadio en las peridotitos, redox del manto y entornos tectónicos: del arcaico al presente».
Earth and Planetary Science Letters 195, 75-90).
[92] Aquí la idea es que, para generar un efecto isotópico independiente de masa a partir de un solo compuesto de partida, por ejemplo el SO
2, que inicialmente presenta una firma isotópica dependiente de masa, se deben generar al menos dos productos que muestren señales isotópicas opuestas. Imaginemos ahora que la subsiguiente química atmosférica, en este caso la oxidación con oxígeno, cause que cada uno de esos productos forme el mismo compuesto, sulfato en este caso. El sulfato se habrá formado combinando las señales isotópicas independientes de masa opuestas de los compuestos originales formados por la fotodisociación del SO
2, generando un producto con la misma señal isotópica dependiente de masa que el SO
2 original.
[93] En realidad, Cloud prefirió el intervalo de edad de 2000 a 2200 millones de años atrás, basándose en las cronologías disponibles en la época. Dataciones posteriores mejoradas han movido las edades un poco hacia atrás.
[94] Dick Holland también examinó la química de los suelos antiguos, los llamados «paleosuelos». Son los remanentes de viejos horizontes de suelo preservados en el registro geológico. Para abreviar, antes del GOE (la gran oxidación), los suelos perdieron una gran cantidad de hierro por erosión química. Esto se debe a que, en una atmósfera baja en oxígeno, el proceso de erosión solubilizaba el hierro de la roca, pero al no haber oxígeno para convertirlo en óxido a partir de la disolución, el hierro disuelto (Fe
2+) era transportado desde el suelo hasta las corrientes y los ríos, y supuestamente hasta los océanos. Después del GOE, el hierro reaccionaba con el oxígeno y así era retenido en el suelo en forma de minerales oxidados (véase, por ejemplo, Rye y Holland, 1998. «Paleosuelos y la evolución del oxígeno atmosférico: una revisión crítica».
American Journal of Science 298, 621-672).
[95] Una de las ideas de Joe Kirschvink es la hipótesis de la «bola de nieve global» (
Snowball Earth). Aquí, Joe reconoció que muchos depósitos glaciales asociados a glaciaciones extendidas durante la era proterozoica estaban localizados cerca del ecuador. Un poco antes, el ruso Mikhail Budyko había hecho un modelo del intercambio de calor en la superficie de la Tierra y había concluido que, si el hielo glacial había llegado alguna vez a 50 grados del ecuador, el albedo habría sido tan alto que la Tierra no hubiera podido retener el calor solar suficiente para impedir que los glaciares cubrieran el planeta. Esto se conoce como glaciación desbocada, y Joe aplicó el modelo a las glaciaciones neoproterozoicas, argumentando que durante esa era la Tierra entró en una glaciación desbocada, posiblemente varias veces, congelándose en una pieza y generando una «bola de nieve global». También hizo algunas sugerencias inteligentes acerca del modo en que la Tierra se escapó de esa situación.
[96] El dióxido de carbono (CO
2), el ácido carbónico (H
2CO
3), el ión bicarbonato (HCO
3-) y el ión carbonato (CO
32-) están relacionados entre sí por un equilibrio químico gobernado por el pH. Al pH del agua marina (alrededor de 8), domina el ión bicarbonato; a un pH más bajo se vuelven más importantes el ácido carbónico y el CO
2; y a un pH más alto gana prominencia el ión carbonato.
[97] Aunque este salto isotópico fue descrito por primera vez por Manfred Schidlowski, del Instituto Max Planck de Química en Mainz, Alemania, en los años setenta.
[98] He tomado este experimento de
Cómo funciona el agua, de Michael Allaby (Dorling Kindersley, Londres, 1995). Llena del todo un vaso transparente con agua coloreada caliente (pero que no hierva) y cubre el vaso con papel de aluminio. Pon el vaso cubierto en un florero, o una pecera, con agua fría sin teñir y retira con cuidado el papel de aluminio, o perfóralo. Y mira lo que pasa. Compáralo con lo que pasa cuando el vaso y la pecera contienen agua a la misma temperatura, o a temperaturas sólo un poco diferentes.
[99] Los planetesimales son objetos sólidos pequeños que se formaron en los primeros estadios de desarrollo del sistema solar. Mediante colisiones violentas, estos planetesimales se fueron agrupando en cuerpos más grandes y, finalmente, en nuestra región del sistema solar, generaron la Tierra.
[100] Representado como la demanda de H
2, que es el doble de la tasa de producción de O
2: 2H
2 + O
2 à 2 H
2O.
[101] Los cálculos de Dick están basados fundamentalmente en el registro isotópico de carbono orgánico y carbono inorgánico, y en lo que ese registro implica sobre las tasas de enterramiento del carbono orgánico a lo largo del tiempo. Para hacer estas estimaciones, también hay que saber cómo ha cambiado con el tiempo el tamaño del reservorio de carbono en la superficie de la Tierra. Estas ideas están tomadas del trabajo de John Hayes y Jake Waldbauer, que publicaron un maravilloso artículo sobre la evolución del ciclo del carbono a lo largo del tiempo (Hayes y Waldbauer, 2006. «El ciclo del carbono y los procesos redox asociados a lo largo del tiempo».
Philosophical Transactions of the Royal Society B 361, 931-950). La idea es que, cuando la Tierra era joven, había menos carbono en la superficie, y las existencias de carbono sólo podían aumentar a medida que el CO
2 era eyectado desde el manto. Hayes y Waldbauer estimaron esta velocidad de crecimiento y los flujos de enterramiento del carbono orgánico a lo largo del tiempo, y Holland usó esos resultados para su modelo.
[102] Lo que es ahora la superficie de la pizarra se encontraba antes en las profundidades de la roca. Cuando las capas superiores se habían erosionado física y químicamente, la superficie que vemos ahora ha quedado expuesta. Pero, en su camino hacia la superficie, esta parte de la pizarra también experimentó la erosión y la exposición al oxígeno aun estando dentro de la roca. Lo que representa hoy la superficie de la roca, por tanto, ha experimentado también todos los estadios intermediarios de la erosión que están experimentando ahora las capas interiores.
[103] Esto fue modelado por John Hayes y Jake Waldbauer, como se discutió en la nota 9 del capítulo anterior.
[104] Esto es una simplificación excesiva, desde luego. Hubo los «tufillos» de oxígeno pre-GOE discutidos en el capítulo 7, y algo de oxidación de la pirita y otras especies sensibles al oxígeno, en aquellos tiempos. Resulta incierto, sin embargo, hasta qué punto fue significativa esta oxidación.
[105] La reacción es:
14H2O + 4FeS2 + 15O2 → 4Fe(OH)3 + 8SO42—+ 16H+.
[106] Y es un argumento algo tautológico, a decir verdad. La evolución de los animales en el precámbrico tardío se toma a veces como una evidencia del incremento de oxígeno en el precámbrico tardío, y a veces la evolución de los animales se atribuye a un incremento de oxígeno en el precámbrico tardío. No puedes apuntarte a las dos cosas mientras no tengas evidencias independientes de un aumento en los niveles de oxígeno en el precámbrico tardío.
[107] Esto tiene que ver con las particularidades del modo en que los reductores de sulfato transportan el sulfato al interior de la célula, y con las rutas bioquímicas que conducen al fraccionamiento dentro de la célula. Así, el fraccionamiento isotópico del azufre ocurre a través de, al menos, dos pasos en el interior de las células, ambos basados en la rotura de enlaces S-O. En el primer paso, el ión sulfato (SO
42-) es reducido a ión sulfito (SO
32-) por la enzima APS reductasa, y en el segundo paso el sulfito es reducido a sulfuro por la enzima sulfito reductasa. El fraccionamiento impuesto por estos pasos, sin embargo, sólo puede detectarse si la célula importa y exporta activamente el sulfato intra y extracelular. De otro modo, todo el sulfato que entra en la célula es reducido a sulfuro y no se observa fraccionamiento alguno, pese a los fraccionamientos dirigidos por enzimas dentro de la célula. Cuando baja la concentración de sulfato, el sulfato se hace limitante para la célula, y se intercambia menos con el entorno extracelular. De este modo, las bajas concentraciones de sulfato conducen a un menor fraccionamiento.
[108] Rob fue parte integral de mi experiencia como estudiante de doctorado. Me visitó cada verano mientras yo trabajaba en mi tesis, y nos hicimos buenos amigos y colegas. Compartimos un montón de risas, un montón de cervezas y un montón de discusiones sobre la geoquímica de los sedimentos marinos modernos y antiguos. Nuestra colaboración se ha mantenido hasta hoy.
[109] La surgencia se refiere al transporte físico del agua de regiones profundas a someras en la columna de agua oceánica. A menudo se estimula por el viento, que puede, por ejemplo, empujar el agua superficial a las lejanías de la costa. Las aguas más profundas ascienden entonces para reemplazar a las aguas superficiales desplazadas. Esto ocurre hoy en muchas regiones del océano, pero de modo más llamativo en las costas occidentales del continente americano.
[110] Dick Holland calculó una vez que se requerirían unos niveles de oxígeno mil veces menores que los actuales para que el Fe
2+ fuera transportado para su deposición en las formaciones de hierro bandeado con 2500 millones de años de Sudáfrica; también estas se formaron en aguas someras.
[111] Estas evidencias fueron aportadas por mi colega Robert Frei, de la Universidad de Copenhague. La versión resumida es que los isótopos de cromo (Cr) se fraccionan durante la erosión oxidativa de los minerales de Cr en tierra firme. Esta erosión oxidativa requiere oxígeno, pero los óxidos de Mn, cuya formación requiere oxígeno, son el oxidante directo. Una vez transportada a los océanos, la señal isotópica del Cr queda fijada en rocas ricas en hierro como las formaciones de hierro bandeado. El registro muestra algunos episodios de Cr fraccionado a partir de 2600 millones de años atrás, lo que ofrece evidencias adicionales de «tufillos» de oxígeno antes del GOE. No hay, sin embargo, evidencias de Cr fraccionado en la formación de hierro Gunflint, de 1880 millones de años de antigüedad, lo que indica unos niveles de oxígeno muy bajos en esa época, que se discutirán en el siguiente capítulo.
[112] De hecho, Lee Kump, de la Universidad Estatal de Pensilvania, ha presentado recientemente evidencias que pueden apoyar esta idea. Lee mostró que la composición isotópica del carbono inorgánico y de la materia orgánica al acabar la anomalía de Lomagundi se volvió demasiado bajo en
13C para poder explicarse por la operación normal del ciclo del carbono, y recurrió a una entrada masiva de carbono bajo en
13C procedente de la oxidación de materia orgánica acumulada en los continentes (Kump, Junium, Arthus, Brasier, Fallick, Melezhik, Lepland, Crne y Luo, 2011. «Evidencia isotópica de la oxidación masiva de materia orgánica tras el Gran Evento de Oxidación».
Science 334, 1694-1696).
[113] Pero no exclusivamente. Algunas de las pizarras más negras que yo he visto provienen de la era neoproterozoica, y la mayoría de ellas contienen muy poca pirita, y probablemente se depositaron bajo unas condiciones ricas en Fe y ferruginosas de la columna de agua.
[114] En la práctica, nos centramos en comprender cómo se repartió el Fe en las muestras, y luego usamos eso para determinar el entorno químico en que se depositaron las muestras. Si las muestras contenían un exceso de Fe respecto a lo esperado en sedimentos normales que se depositan desde una columna de agua rica en oxígeno, eso significa que la columna de agua sobre el sedimento estaba libre de oxígeno, y por tanto capaz de transportar Fe
2+ disuelto a través del agua y finalmente hasta el sedimento. Si la mayoría del enriquecimiento en Fe estaba contenido en pirita, eso significa que la columna de agua contenía sulfuro. Si el Fe enriquecido era pobre en pirita, entonces la concentración de sulfuro era baja, y la columna de agua contenía Fe
2+. La columna no podía ser a la vez rica en sulfuro y en Fe
2+, debido a la baja solubilidad de los minerales de sulfuro de hierro.
[115] ¡Ostras, otro sistema de isótopos! Como vimos en el capítulo 7, el molibdeno (Mo) se libera durante la erosión oxidativa en tierra firme y luego se transporta por los ríos al océano. Hay dos rutas principales de retirada en el océano. Una es por adsorción en óxidos de Fe y Mn, lo que representa la ruta de remoción óxica, y aquí se provoca un gran fraccionamiento. Hay, sin embargo, muy poco fraccionamiento durante la remoción en entornos sulfurosos, que es la segunda ruta de retirada. Por tanto, los sedimentos depositados en escenarios sulfurosos aportan una estimación de la composición isotópica del Mo en el agua marina primitiva, y esta composición isotópica aporta una medida de la proporción entre las rutas de remoción óxica y sulfurosa. De manera que Gail Arnold y sus colegas presentaron evidencias, basadas en los isótopos del Mo, de una ruta sulfurosa reforzada para la remoción del Mo en sedimentos de 1600 millones de años del norte de Australia, que se depositaron en un escenario sulfuroso.
[116] El Fe
2+ habría venido de una combinación de fuentes hidrotermales de fumarolas de los fondos oceánicos y de ríos en los que gran parte del hierro habría estado inmovilizado como partículas fluviales recubiertas de óxidos de hierro. Estos óxidos de hierro inmovilizados pueden reducirse al estado ferroso soluble en condiciones anóxicas.
[117] Carta al rey Carlos I, 19 de agosto de 1628. Manuscrito CO 1/5 (27), 75, MHA 16-B-2-011, trascrito por P. E. Pope, con ortografía y puntuación modernizada. De la Oficina de Registro Público, Oficina Colonial, Londres, Inglaterra, 1999. Documentos relacionados con Ferryland: 1597-1726. Historia de la Colonia de Avalon, Fundación de la Colonia de Avalon, Ferryland, NL, y Proyecto de sitio web del patrimonio de Terranova y Labrador, Universidad Memorial de Terranova, St. John’s, NL, Canadá.
[118] Las células epiteliales recubren las superficies y las cavidades de los animales y actúan como cubierta protectora, facilitan el intercambio y permiten a los organismos percibir su entorno.
[119] La fauna de Ediacara, aunque muy visible y fácilmente reconocible en el registro fósil, tal vez no represente la evidencia más antigua de vida animal fósil. Gordon Love, de la Universidad de California en Riverside, ha identificado biomarcadores asociados a las esponjas en rocas de unos 635 millones de años de edad (Love y col., 2009. «Los esteroides fósiles registran la aparición de las demoesponjas durante el periodo criogénico».
Nature 457, 718-721); y Adam Maloof, de la Universidad de Princeton, ha encontrado algunos posibles cuerpos fósiles de esponjas en rocas ligeramente más antiguas (Maloof y col., «Posibles fósiles de cuerpos de animales en calizas pre-Marinoan del sur de Australia».
Nature Geoscience 3, 653-659).
[120] La glaciación Gaskiers fue posiblemente la última, y quizá también la menor, de las grandes glaciaciones que puntuaron la segunda mitad de la era neoproterozoica. Algunas de estas glaciaciones fueron tan grandes que casi envolvieron en hielo la Tierra, generando la llamada bola de nieve planetaria (
Snowball Earth). La hipótesis de la bola de nieve planetaria es una creación de Joe Kirschvink; conocimos a Joe en el capítulo 8. La hipótesis de la bola de nieve planetaria se discute con más detalle en la nota 3 del capítulo 8, y se hizo célebre gracias a Paul Hoffman y sus colegas en un extraordinario artículo de 1998 (Hoffman, Kaufman, Halverson y Schrag, 1998. «Una bola de nieve planetaria en el neoproterozoico».
Science 281, 1342-1346). Estas glaciaciones y su potencial influencia en la vida y en la química global se han discutiso activamente en la literatura, pero se ha alcanzado muy poco consenso. He decidido no centrarme aquí en esas glaciaciones, pero se puede encontrar una buena introducción en el libro de Andy Knoll
La vida en un planeta joven (Princeton University Press, Princeton, NJ, 2003).
[121] Recolectamos con las autorizaciones adecuadas, ¡y no de los famosos yacimientos de fósiles!
[122] En un contexto geológico, «océano profundo» significa normalmente cualquier cosa por debajo de la base de las olas de tormenta, o por debajo de unos 100 metros de profundidad. Lo que no es tan profundo, realmente. En el caso de las rocas de Avalon, evidencias independientes indican que probablemente estamos examinando unas profundidades mucho mayores: cientos de metros, o quizá incluso un kilómetro. A menos que tengamos un trozo de suelo oceánico profundo situado (obducido) en los continentes, nunca podemos examinar al océano realmente profundo.
[123] Los otros científicos que contribuyeron a este trabajo incluyen a Andy Knoll, Guy Narbonne, Gerry Ross, Tatiana Goldberg y Harald Strauss.
[124] La primera evidencia no debe confundirse con la primera aparición. Podría haber episodios anteriores de oxigenación del océano profundo que se nos han pasado.
[125] En el mundo moderno, el agua totalmente saturada de aire contiene una cantidad de oxígeno que depende de la temperatura y la salinidad del agua. El agua del fondo del océano se deriva de regiones polares donde la concentración saturada de aire es de 325 micromoles por litro. El agua de lagos y ríos, y la que sale del grifo en casa, probablemente no están muy lejos de ese valor.
[126] En una contribución anterior, Andreas Teske, ahora en la Universidad de Carolina del Norte, y yo mismo llegamos a una estimación similar de los niveles mínimos de oxígeno. Nos basamos en la cantidad de oxígeno que se requiere para oxigenar los sedimentos de la superficie marina, ya que mostramos que un ciclo oxidativo del azufre estaba activo en los sedimentos, y que requería oxígeno.
[127] Si rompes un papel en dos, el área junto a los márgenes del papel se incrementará. Si vuelves a romper en dos cada trozo, el área se incrementará más aún.
[128] Lou examinó la fracción de
87Sr/
86Sr en el estroncio preservado en las calizas. Si procesos posteriores no la han alterado, la composición isotópica del Sr en el agua del mar, que es incorporada en las calizas mientras precipitan, indica el balance entre la entrada de estroncio desde los continentes, que tiene una fracción alta de
87Sr/
86Sr, y la entrada desde las fumarolas hidrotermales, que tiene una fracción baja de
87Sr/
86Sr. La fracción de las calizas se incrementa drásticamente alrededor de 600 millones de años atrás, indicando una mayor entrada de productos de erosión continentales al océano, y por tanto una tasa alta de enterramiento de sedimentos con materia orgánica asociada. Lou, sin embargo, no construyó un modelo del oxígeno, y una de mis objeciones es que una tasa alta de consumo de oxígeno asociada con tasas altas de erosión continental habría equilibrado las altas tasas de liberación de oxígeno asociadas con las tasas altas de enterramiento de carbono. Esta puntualización fue avanzada por Bob Berner, y la exploraremos en más detalle en el capítulo 11.
[129] Hay científicos que no aceptarán esto, entre ellos Martin Kennedy, de la Universidad de Adelaida, y también Lou Derry. Arguyen que la anomalía es un efecto diagenético causado por la percolación de fluidos a través de las calizas mucho después de la deposición original, que habría cambiado las composiciones isotópicas originales. No se puede descartar que los efectos diagenéticos hayan alterado las calizas, y en particular sus composiciones de
18O. Sin embargo, la señal de
13C se observa en muchos lugares del mundo, y puede extenderse a lo largo de cientos de kilómetros en una sola localización. A mí no se me ocurre ningún mecanismo diagenético plausible que pueda producir unos rasgos de
13C de semejante escala y tan reproducibles, y por tanto prefiero la interpretación de que representan las composiciones originales del agua marina.
[130] Nosotros argumentamos que la liberación y oxidación de un reservorio gigante de metano retenido en los sedimentos como fases de hidrato de clatrato podía ser una mejor solución. Estas fases se encuentran hoy en los sedimentos marinos, y están incluso más mermados en
13C que el carbono orgánico disuelto (DOC) que se halla en los océanos.
[131] Se necesita un menor sumidero de oxígeno para producir la anomalía de Shuram-Wonoka por oxidación de metano que para producirla por oxidación de DOC. La razón es que el metano está mucho más mermado en
13C que el DOC marino. De hecho, Christian Bjerrum y yo modelamos la anomalía con oxidación de metano preservando el suficiente oxígeno para la respiración de los animales primitivos.
[132] Claramente, la evolución primaria de los animales primitivos ocurrió mucho antes de esto, y probablemente no fue influida por las elevadas demandas de oxígeno de los organismos motiles. De hecho, unos resultados preliminares de nuestro grupo indican que el 2% de los niveles actuales de oxígeno puede ser suficiente para la respiración de las esponjas, que son las formas más basales de los animales modernos en el árbol de la vida.
[133] En realidad sí hay ventanas, pero son unas pequeñas ranuras que obedecen más a efectos de diseño que a la intención de dar a la gente una sensación de la luz y la vida que hay fuera del edificio.
[134] También hemos visto esta capacidad en Vladimir Vernadsky, Preston Cloud y Dick Holland, todos ellos mencionados anteriormente en el texto.
[135] No entraré en los detalles de esto, pero las tasas de erosión deberían depender directamente del área continental disponible para la erosión. Esas tasas deberían depender también de la actividad del ciclo hidrológico y de la temperatura, con mayores tasas de erosión a mayor temperatura. En este caso, más lluvia conduce a una erosión más rápida. Cuanto mayor sea el relieve de los continentes, es decir, la altura de las montañas respecto a los valles, más intensa será la erosión, sobre todo la erosión física. También las plantas y sus raíces perturban las rocas y los suelos, y elevan los niveles de CO
2 en el suelo; estas influencias se combinan para incrementar las tasas de erosión.
[136] Ronov fue una gran inspiración para la comunidad geológica occidental, incluso durante la era soviética, cuando el contacto entre los científicos occidentales y soviéticos era difícil en el mejor de los casos.
[137] Son bajos en azufre porque las concentraciones de sulfato son bajas en las aguas terrestres como lagos y ríos. Las bajas concentraciones de sulfato limitan la magnitud de la reducción de sulfato, y por tanto la cantidad de sulfuro que se puede producir para reaccionar ya sea con el hierro para formar pirita, o con la materia orgánica para formar compuestos orgánicos azufrados.
[138] La tasa de deposición de un tipo concreto de sedimento se calcula como la proporción del sedimento total que representa un tipo de roca individual multiplicado por la tasa total de deposición de sedimentos; por ejemplo, si el carbón depositado durante un tiempo dado representa el 10% del sedimento total, el multiplicador es 0,1.
[139] Esto se debe a que la erosión del carbono orgánico y el azufre pirítico, y también su enterramiento subsiguiente otra vez en sedimentos marinos, están acoplados a la tasa total de deposición de sedimentos. Los procesos están acoplados porque la erosión en tierra genera el sedimento que se deposita en el mar. Más aún, las tasas de enterramiento del carbono orgánico y el azufre pirítico son una función lineal de la tasa de sedimentación. Así, puesto que tanto la liberación como el consumo de oxígeno vienen gobernados por la tasa total de deposición de sedimentos, la variabilidad de esta tasa tiene poca influencia en la acumulación neta o en el consumo de oxígeno atmosférico. Véase la nota 12 del capítulo anterior.
[140] También hay una similitud entre las tasas calculadas de enterramiento de azufre a partir de los datos de abundancia de sedimentos y las calculadas a partir de los isótopos del azufre.
[141] Lovelock introdujo la hipótesis de Gaia en los años setenta (Lovelock y Margulis, 1974. «Homeostasis atmosférica por y para la biosfera: la hipótesis de Gaia». Serie
Tellus A 26, 2-10; y Lovelock, 1979.
Gaia: una nueva mirada a la vida en la Tierra, Oxford University Press, Oxford) y, en su forma original, Lovelock concibió una biosfera global que actuaba en armonía para generar un entorno químico optimizado para la vida. De este modo, la vida controlaba el entorno. Cuando fue resultando evidente que la química de la superficie de la Tierra ha cambiado con el tiempo, la idea de una optimización invariante se volvió más relajada, pero la idea de que la vida ha contribuido con fuerza a dar forma, o incluso a controlar, el entorno químico ha perdurado. En verdad, la hipótesis de Gaia tiene mucho en común con los escritos anteriores de Vladimir Vernadsky, como se discutió en el capítulo 6. Pocos geobiólogos en activo negarían la importancia de la vida para influir en el entorno químico. El papel de la vida en la regulación activa de esta química es un área de investigación en la frontera del conocimiento, y un tópico de intensa discusión.
[142] Una de estas formas es a través de la fotorrespiración. Si recordamos el capítulo 3, la enzima Rubisco es responsable de la fijación de carbono en el ciclo de Calvin, y también tiene actividad oxigenasa (consumiendo O
2 y liberando finalmente CO
2), lo que compite directamente con su función carboxilasa (que fija CO
2 para formar carbono orgánico). Esto significa que la enzima se vuelve menos eficiente fijando carbono a niveles altos de oxígeno, puesto que la actividad oxigenasa adquiere más importancia. Esto se puede ver como una retroalimentación negativa biológica de nivel organísmico sobre el incremento de oxígeno, aunque la mayor parte de las evidencias indican que la actividad de oxigenasa es más una extravagancia evolutiva que una decidida innovación biológica para la estabilización del oxígeno. En cualquier caso, Bob reconoció su importancia como un regulador del oxígeno y como un mecanismo de retroalimentación en su modelo del oxígeno.
[143] Si retrocedemos al capítulo 1, la luminosidad solar fue importante al considerar la regulación de la temperatura en la Tierra joven; es menos significativa en el eón fanerozoico, aunque todavía tiene alguna importancia.
[144] Estas curvas de isótopos se pueden recuperar porque el modelo COPSE computa los antiguos ciclos del carbono y el azufre, incluidas las tasas de enterramiento del carbono orgánico y la pirita en los sedimentos. Así, la composición isotópica de todos los elementos de los antiguos ciclos del carbono y el azufre se puede predecir.
[145] Puede haber también otra forma de mirar a esto. Como vimos en el capítulo 5, la incidencia de incendios forestales es sensible al oxígeno. La combustión experimental de material vegetal, acoplada con el modelado, sugiere que las plantas no arderán a niveles de oxígeno por debajo del 16%, y que la combustión se vuelve rampante por encima del 22% (la atmósfera actual es 21% O
2) (Belcher, Yearsley, Hadden, McElwain y Rein, 2010. «Inflamabilidad intrínseca de fondo de los ecosistemas terrestres estimada a partir del oxígeno paleoatmosférico a lo largo de los últimos 350 millones de años».
PNAS 107, 22448-22453). El carbón se forma cuando arden las plantas, y el registro geológico de los restos de carbón indica que los incendios ocurrían ya tan pronto como en el silúrico tardío, hace unos 418 millones de años (Scott y Glasspool, 2006. «La diversificación de los sistemas de fuego paleozoicos y las fluctuaciones de la concentración de oxígeno atmosférico».
PNAS 103, 10861-10865). Unos niveles elevados de oxígeno del 16% en esa época serían compatibles con las evidencias de isótopos del molibdeno, y coherentes con el modelo Geocarbsulf. El modelo COPSE predice una elevación algo más tardía del oxígeno a esos niveles, pero se le podría hacer consistente con los resultados del carbón haciendo unas suposiciones diferentes sobre el tiempo de desarrollo de los ecosistemas de plantas terrestres.
